
Фенилкетонурия у детей: особенности лечения и рекомендации врачей

- Механизм формирования
- Основные виды
- Причины
- Симптомы
- Диагностика
- Лечение
- Особенности питания
- Осложнения и прогноз

Фенилкетонурия у детей — наследственное заболевание, обусловленное мутацией гена в 12 хромосоме
Механизм формирования
Фенилкетонурия у детей (ФЛК) – наследственно обусловленная болезнь, связанная с нарушением синтеза, распада и эвакуации продуктов аминокислотного обмена из организма, а именно — преобразованием фенилаланина до тирозина. Таким образом, происходит накопление тех веществ, которых в организме быть не должно – ортофенилацитата, фенилмолочной и фенилпировиноградной кислот, фенилэтиламина. Высокие концентрации этих веществ, подобно яду влияют на организм, приводя к следующим состояниям:
- нарушению жирового обмена в головном мозге;
- гибели нейромедиаторов, которые передают импульсы между мозговыми клетками;
- общей интоксикации.
Мишенью при фенилкетонурии является нервная система, поэтому интоксикация приводит к выраженному снижению интеллекта и умственной отсталости — олигофрении.
Заболевание возникает, если носителем мутантного гена являются оба родителя. При этом они могут сами не болеть патологическим синдромом. Риск мутации у ребенка в таком случае достигает 25-30%.
Основные виды
Клиницисты выделяют 3 основных типа патологического состояния у детей:
- I тип. Составляет 95% всех случаев детской фенилкетонурии, обусловлен нарушением синтеза фениланин-4-гидроксилазы.
- II тип. Состояние характеризуется невозможностью выработки фермента дигидроптеридинредуктазы. Возникает в 1-3% среди пациентов с ФЛК. Лечение затруднено, а при его отсутствии наблюдается высокая летальность детей уже в 2-3 года.
- III тип. На фоне патологии наблюдается нарушение синтеза еще одного фермента — тетрагидробиоптерина. Редкая разновидность.
Существуют еще несколько типов заболевания, но они составляют всего 0,1-0,5% от общего числа выявленных случаев.
Причины
Фенилкетонурия характеризуется аутосомно-рецессивным путем наследования. При появлении характерных признаков болезни говорят о мутации гена, который напрямую отвечает за выработку фенилаланина и 12 хромосомы. Отсутствие прямых признаков указывает на развитие ФЛК 2 и 3 типов.
Спровоцировать заболевание способны геномные мутации вблизи 12 хромосомы, перенесенный родителями полиомиелит в раннем детстве, браки между близкими родственниками, системный токсический фактор (например, радиация), муковисцидоз в анамнезе. Повреждение гена может носить спонтанный характер.
Симптомы

Питание — важное условие для поддержания адекватного качества жизни
Сразу после рождения ребенка симптоматический комплекс не выражен или не проявляется вовсе. Первые признаки наблюдаются спустя 2-3 недели или месяц после рождения малыша.
В первые несколько недель жизни заболевание не дает о себе знать, а дети не отличаются от своих сверстников. Начальные признаки нарушения возникают позднее. Среди клинических проявлений у новорожденных выделяют судороги, повышенный мышечный тонус, апатичность, обильные срыгивания, рвоту.
Наиболее характерными для фенилкетонурии считаются следующие поздние признаки:
- заметная и нарастающая умственная отсталость (малыш не интересуется окружающим миром, не узнает окружающих, в позднем возрасте отсутствует способность к обучению, а полученные навыки быстро утрачиваются);
- неприятный запах от кожи (у больных фенилкетонурией наблюдается характерный запах плесени от кожных покровов);
- дерматологические заболевания;
- поздний рост молочных зубов;
- заметное отставание в физическом развитии.
Дети с ФЛК имеют типичную внешность: кожа без пигментации, бледная, глаза и волосы светлые из-за нарушения выработки меланина. Нередко наблюдаются бесконтрольные телодвижения, как при ДЦП.
Диагностика
Фенилкетонурия является областью исследования генетиков, психиатров, невропатологов и гепатологов. Окончательный диагноз ставят на основании следующих данных:
- обнаружение в крови фенилаланина в количестве более 900 мгмоль/л;
- положительный результат пробы Феллинга;
- присутствие в крови фенилмолочной, фенилуксусной или фенилпировиноградной кислоты.
Важными критериями диагностики фенилкетонурии у детей являются жалобы родителей на нетипичное поведение, задержку психофизического развития ребенка, прочие частые при ФЛК симптомы.
Высокой достоверностью обладает ранняя диагностика заболевания, еще в период внутриутробного развития плода. Сегодня каждая женщина, наблюдающаяся у гинеколога по поводу беременности, проходит ряд скрининговых обследований, в том числе и на фенилкетонурию.
Лечение
Лечение фенилкетонурии только консервативное. Оно направлено на стабилизацию метаболических процессов, купирование симптомов, предупреждение олигофрении и других психических расстройств личности. Обязательно назначаются витаминные комплексы, минеральные соединения, лечебная гимнастика, массаж, иглорефлексотерапия.
По показаниям обязателен прием ноотропных, противосудорожных или антиконвульсивных препаратов. При необходимости ребенок наблюдается у дефектолога, логопеда, проводится регулярный анализ крови и электроэнцефалограмма.
Важную роль в терапии фенилкетонурии играет лечебное питание. Учитывая, что фенилаланин проникает в организм с пищей, то коррекция состояния предполагает правильную диету. Основу рациона составляют фрукты, овощи, аминокислотные смеси, гидролизаты. Расширение рациона допустимо только после совершеннолетия, когда наблюдается терпимость организма к фенилаланину.
Особенности питания

Признаки заболевания носят неврологический характер, определяют умственно-физические критерии развития ребенка
Вопрос диеты встает остро уже с самого рождения ребенка. С каждым месяцем при отсутствии терапии и корректирующей диеты наблюдается снижение интеллекта на 3-4 балла по шкале IQ. При уровне фенилаланина в крови до 6% от нормы питание не корректируют. При превышении показателей переходят на безбелковые смеси. Прикорм начинают с овощей, фруктов и соков на их основе. Позже вводят фруктовое пюре из груш, яблок, слив, овощное пюре.
Далее в рацион вводят безмолочные каши, низкобелковые смеси, безбелковый хлеб. Лечебная диета носит пожизненный характер, однако небольшие погрешности допустимы по достижении ребенком совершеннолетия.
Диета при фенилкетонурии исключает следующие продукты:
- птицу, мясо, бульоны на их основе;
- молочные продукты и продукцию на основе молока, включая грудное молоко;
- любую рыбу.
По мере взросления количество суточного белка ребенку увеличивают, но постоянно наблюдают за содержанием фенилаланина в крови. Питание строго контролируют, меню составляют в соответствии с данными энцефалографии, анализа крови. Наряду с повышением суточного белка снижают количество жиров и углеводов. При необходимости получения клинических рекомендаций по поводу лечебного рациона можно обратиться к врачу-гастроэнтерологу, диетологу.
При отмене или несоблюдении правил лечебного питания наблюдается возобновление типичной симптоматики даже на фоне медикаментозной терапии, нарастание умственной отсталости. Диета направлена на поддержание лабораторных показателей в норме, чтобы исключить необратимые изменения в головном мозге.
Осложнения и прогноз
При несоблюдении корректного рациона прогноз по заболеванию крайне неблагоприятный. Ребенок становится умственно отсталым, инвалидом уже через несколько лет. При начале терапии спустя пару лет после рождения могут наблюдаться остаточные расстройства психики, связанные с гибелью клеток головного мозга. Родителям важно соблюдать все клинические рекомендации, объяснять подросшему ребенку опасность несоблюдения диеты.
Массовый скрининг на фенилкетонурию помогает родителям и врачам выстроить адекватную, своевременную тактику ведения больного ребенка, обеспечив ему благоприятные прогнозы по развитию в будущем.
Что такое фенилкетонурия?
Фенилкетонурия (ФКУ) — это наиболее распространенное врожденное нарушение метаболизма аминокислот, возникающее в результате дефицита фермента фенилаланингидроксилазы. Вследствие данного заболевания снижается способность организма к метаболизму аминокислоты фенилаланина. Эти процессы вызывают накопление фенилаланина в тканях организма и физиологических жидкостях.
Повышенные уровни фенилаланина негативно влияют на когнитивные функции, поэтому у людей с классической фенилкетонурией почти всегда присутствует интеллектуальная инвалидность. Заболевание доходит до этой стадии, если уровни фенилаланина не контролируются путем диетической или медикаментозной терапии.
Механизм развития и причины фенилкетонурии
Механизм, вследствие которого повышенные уровни фенилаланина вызывают интеллектуальную инвалидность, в точности не известен, однако ограничение продуктов, содержащих вредные вещества, снижает этот эффект.
В детском возрасте уровень фенилаланина в крови и коэффициент интеллекта (IQ) тесно связаны.
В настоящее время в ведущих клиниках мира ведется детальное изучение незначительных нейропсихологических дефицитов у детей с контролируемой фенилкетонурией. Некоторые ученые списывают этот дефицит на остаточные аномалии функции нейромедиаторов — например, снижение производства нейротрансмиттеров в результате недостаточного транспорта тирозина сквозь нейронную клеточную мембрану.
Иногда уровни аминокислот у больных в норме, но присутствует дефицит синтеза или утилизации белка BH4. Такое состояние называется злокачественной фенилкетонурией. BH4 кофактор необходим для гидроксилирования тирозина (предшественника дофамина) и триптофана (предшественника серотонина). Таким образом, пациенты с дефицитом BH4 могут иметь дополнительные проблемы неврологического характера, которые невозможно полностью скорректировать диетическим ограничением количества фенилаланина. Таким пациентам необходимы дополнительные методы лечения, которые не гарантируют стопроцентной эффективности.
Фенилкетонурия — это аутосомно-рецессивное заболевание, вызванное мутацией в гене ФАГ (фенилаланингидроксилазы). На сегодняшний день учеными идентифицировано более 500 различных мутаций гена ФАГ.
Для фенилкетонурии характерна заметная генотипическая гетерогенность, как внутри определенной популяции, так и между различными популяционными группами населения. Генотип и фенотип людей с данным заболеванием тесно связаны, кроме того, неродственные индивидуумы с одинаковыми мутациями имеют определенную степень изменчивости толерантности к фенилаланину. Генотип — это совокупность наследственных генетических характеристик, а фенотип — это совокупность внешних факторов среды, влияющая на человека уже после рождения.
ФКУ является наследственным заболеванием, вызванным дефектом в гене ФАГ. Ген ФАГ помогает создать фенилаланингидроксилазу — фермент, ответственный за разрушение фенилаланина. Опасное накопление фенилаланина может произойти, если человек употребляет чрезмерное количество продуктов с высоким содержанием белка, например, яйца и мясо. Оба родителя должны иметь дефектную версию гена ФАГ, только тогда ребенок наследует расстройство. Если только один родитель является носителем измененного гена, ребенок не будет иметь каких-либо симптомов, но так же будет являться носителем гена.
Диагностика фенилкетонурии
С 1960-х годов в больницах Соединенных Штатов был введен регулярный скрининг новорожденных на ФКУ путем взятия пробы крови. В настоящее время такая диагностика всё еще актуальна. Проводится взятие пробы крови из пятки новорожденного, а затем анализируется на ФКУ и другие генетические нарушения.
Скрининг-тест выполняется, когда новорожденному исполняется один-два дня, и он еще находится в роддоме.
Дополнительные тесты могут быть выполнены, чтобы подтвердить первоначальные результаты. Обычно врачи проверяют ген ФАГ на наличие мутаций, которые вызывают фенилкетонурию.
Если ребенок или взрослый проявляет симптомы фенилкетонурии, например, очевидна задержка развития, необходимы дополнительные анализы крови для подтверждения диагноза. Тест подразумевает взятие образца крови и ее анализ на присутствие фермента, необходимого для расщепления фенилаланина.
Симптомы фенилкетонурии
Большинство детей с фенилкетонурией кажутся нормальными при рождении. Если анализ крови после рождения не был проведен, а заболевание осталось невыявленным, происходит прогрессивная задержка развития — наиболее распространенный симптом фенилкетонурии.
Первые симптомы фенилкетонурии у новорожденных:
- рвота;
- судороги;
- неприятный запах от тела, так называемый «мышиный» запах (из-за выделения фенилуксусной кислоты);
- экзема;
- микроцефалия;
- расстройства поведения (раздражительность, плаксивость, отказ от груди). Ребенок плохо спит, его поведение отличается особым беспокойством. Такие дети по мере взросления становятся буквально неуправляемыми,
У пожилых людей с фенилкетонурией на МРТ отчетливо видны проявления демиелинизации. Присутствует заторможенность когнитивных функций и двигательных навыков. Коэффициент умственного развития снижается на 10 единиц и более, если прекратить придерживаться рекомендуемой врачом диеты.
Симптомы (кожа):
- изменение цвета волос и оттенка кожи, являющееся следствием ухудшения синтеза меланина.
Больной имеет волосы светло-рыжего или белого цвета и болезненно бледную кожу, голубые глаза, светлые ресницы и брови.
Такое изменение цвета волос и кожи может быть характерным даже для афроамериканцев и азиатов, однако это скорее редкость, чем норма.
Другие симптомы кожных проявлений фенилкетонурии:
- экзема (атопический дерматит);
- увеличение числа гнойных инфекций;
- фолликулярный кератоз;
- потеря волос.
Симптомы фенилкетонурии могут быть как легкими, так и интенсивными. Наиболее тяжелой формой расстройства является так называемая классическая фенилкетонурия. Ребенок с классическим типом заболевания может казаться абсолютно нормальным первые несколько месяцев жизни. Если заболевание не выявляется и не лечится, появляются первые симптомы, которые затем усиливаются.
К таким симптомам относятся:
- аномалии функции глаз;
- паркинсонизм;
- гипопигментация кожи;
- «мышиный» запах, характерный для мочи и пота;
- интеллектуальная инвалидность;
- эпилепсия;
- тремор конечностей;
- задержка роста;
- гиперактивность;
- кожные заболевания;
- неприятный запах изо рта;
- неприятный запах изо рта;
Менее тяжелая форма фенилкетонурии — это состояние, при котором у новорожденного в организме слишком большое количество фенилаланина. Дети с этой формой заболевания могут иметь только легкие симптомы, но они должны придерживаться специальной диеты, чтобы избежать развития умственных нарушений.
Если фенилкетонурия не диагностируется при рождении и лечение не начинается быстро, расстройство может вызвать:
- необратимое повреждение мозга и психические расстройства в течение первых нескольких месяцев жизни;
- поведенческие проблемы и судороги у детей более старшего возраста.
После того, как больные начинают соблюдать специфическую диету и другие необходимые процедуры, интенсивность симптомов начинает уменьшаться. При строгом контроле рациона питания симптомы заболевания отсутствуют.
Лечение фенилкетонурии
Пациенты с фенилкетонурией могут облегчить тяжесть симптомов и предотвратить развитие осложнений, следуя специальной диете и принимая лекарства.
Диета
Самой эффективной диетой является ограничение продуктов, содержащих фенилаланин. Дети с ФКУ не могут потреблять грудное молоко, они питаются по специальной схеме, употребляя продукт Лофеналак или его аналоги — продукты, специально предназначенные для людей с таким заболеванием.
Если ребенок достаточно взрослый, чтобы жевать твердую пищу, необходимо полностью исключить такие продукты:
- яйца;
- сыр;
- орехи;
- молоко;
- фасоль;
- курицу;
- говядину;
- свинину;
- рыбу.
Для того чтобы больные могли получать достаточное количество белка, дети с ФКУ употребляют специальные пищевые добавки, содержащие все аминокислоты, необходимые организму, за исключением фенилаланина.
Существуют также определенные продукты с низким содержанием белка, которые можно найти в специализированных магазинах или аптеках. Эти пищевые добавки больные должны употреблять всю жизнь, чтобы избежать снижения умственных способностей и появления различных неприятных симптомов.
Все пациентам с фенилкетонурией необходимо постоянно контактировать с диетологом или лечащим врачом, чтобы правильно поддерживать надлежащий баланс питательных веществ, ограничивая при этом потребление фенилаланина. Также необходимо контролировать уровни фенилаланина путем ведения учета количества фенилаланина в продуктах питания, которые человек потребляет в течение дня.
Медикаментозное лечение
Управление качеством пищевых продуктов и медикаментов США (FDA) недавно одобрило препарат Сапроптерин (Kuvan) для лечения фенилкетонурии. Sapropterin помогает снизить уровень фенилаланина. Этот препарат должен быть использован в сочетании со специальной схемой употребления пищи. Однако наиболее эффективен он для людей с легкими формами фенилкетонурии.
Беременность и фенилкетонурия
Женщина с фенилкетонурией в период беременность подвергается риску осложнений, если не следует схеме питания, рекомендованной врачом. Среди возможных рисков следует отметить вероятность выкидыша. Также есть шанс, что пока еще нерожденный ребенок будет подвергаться воздействию высоких уровней фенилаланина. Это может привести к различным проблемам у плода, в том числе:
- ограниченным интеллектуальным возможностям;
- порокам сердца;
- задержке роста;
- низкому весу на момент рождения;
- аномально маленькой голове (микроцефалии).
Все эти признаки становятся заметными у новорожденного не сразу, но тест, проведенный в первые сутки – двое после рождения, может помочь определить наличие заболевания.
Иногда лечение больных с фенилкетонурией (ФКУ) проводят в специальных клиниках, профилем которых являются метаболические нарушения. Обычно схема лечения разрабатывается комплексно, при участии эндокринолога, иммунолога и диетолога.
Уровни фенилаланина следует проверять через регулярные промежутки времени, примерно 1-2 раза в неделю у новорожденных и один раз в месяц для детей старшего возраста и взрослых.
Большинство американских клиник рекомендуют удерживать уровень фенилаланина в пределах 2-6 мг/дл (120-360 мкмоль/л). Точное попадание в рекомендуемый диапазон показателей требует квалифицированного ухода и тщательного мониторинга.
Диету ни в коем случае не рекомендуется прекращать по достижении подросткового возраста, поскольку гиперфенилаланинемия может иметь пагубные последствия и для взрослых пациентов.
Некоторые взрослые пациенты с незарегистрированной фенилкетонурией и, следовательно, интеллектуальной инвалидностью, могут значительно улучшить когнитивные показатели и физическое состояние при соблюдении специальной диеты.
Больные, проходящие качественное лечение, могут придерживаться нормального уровня активности.
Прогноз при фенилкетонурии
Прогноз для разных групп пациентов сильно отличается. Например, дети, заболевание которых было обнаружено в раннем возрасте (в течение первого месяца жизни), при тщательном контроле состояния здоровья, соблюдении режима питания, могут жить абсолютно нормальной жизнью.
Если ребенок не придерживается диеты или соблюдает её частично, нарушения умственного развития неизбежны.
Большинство больных с фенилкетонурией, не имеющих никакого лечения, принадлежат к категории людей с глубокой интеллектуальной инвалидностью.
Распространенные трудности таких больных это:
- психологические проблемы, например, агорафобия и другие фобии;
- нарушение внимания;
- пониженная способность к концентрации;
- нарушения поведения.
Долгосрочный прогноз для пациентов с ФКУ очень хороший, если сразу после рождения ребенка родители придерживаются специальной диеты. Интеллектуальная инвалидность без лечения наступает уже в первый год жизни.
Проявляется она в следующем:
- заторможенность;
- нарушения поведения;
- неспособность к адекватному выражению эмоций (ребенок не узнает родителей, капризничает и т.д.);
- неврологические проблемы (судороги, тремор).
Можно ли предотвратить фенилкетонурию?
ФКУ является генетическим заболеванием, поэтому она не может быть предотвращена. Однако, ферментный анализ — это один из способов узнать, являются ли муж или жена носителем дефектного гена.
Всем больным с ФКУ следует избегать аспартама (искусственного подсластителя). Аспартам широко используется в производстве лекарственных средств, витаминов, напитков и других съедобных продуктов.
Диета с исключением фенилаланина увеличивает вероятность метаболических проблем, например, снижает способности организма к усвоению фолиевой кислоты или жирных кислот.
По материалам:
© 2005 — 2016 Healthline Media.
© 1998-2016 Mayo Foundation for Medical Education and Research.
Georgianne L Arnold, MD; Chief Editor: Luis O Rohena, MD.
Фенилкетонурия ( Болезнь Феллинга , Фенилпировиноградная олигофрения )
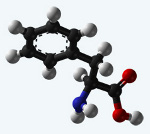
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
МКБ-10
- Причины фенилкетонурии
- Патогенез
- Симптомы фенилкетонурии
- Диагностика
- Лечение фенилкетонурии
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
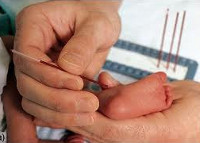
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
- Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
- Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
- Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
- Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Фенилкетонурия — что это такое? Симптомы, диагностика и лечение. Как наследуется фенилкетонурия
Сейчас диагностируется огромное количество наследственных заболеваний, которые ребенок получает от отца или от матери. Экологическая обстановка, неправильное питание, нездоровый образ жизни – все это приводит к тому, что клетки мутируют, генетическая информация претерпевает существенные изменения. Вот от этого и возникает огромное количество наследственных заболеваний. Одним из них является фенилкетонурия. Что это за болезнь, знают не многие, поэтому постараемся во всем разобраться.
Сущность понятия
Фенилкетонурия является наследственным заболеванием, связано оно с серьезными нарушениями в белковом обмене. Это, в свою очередь, приводит к поражению нервной системы.
Неработоспособность всего одного фермента, фенилаланина, а в результате – такие серьезные проблемы со здоровьем, как фенилкетонурия. Что это за состояние, когда в организме происходит накопление большого количества токсических веществ? Все ядовитые соединения запасаются в биологических жидкостях, поэтому для медиков диагностировать болезнь обычно не составляет особого труда.
Если вовремя не принять меры, то можно наблюдать серьезные поражения нервной системы, а это уже приводит к нарушениям в функционировании всего организма.
Таким образом, без соответствующего лечения о нормальной жизни пациента не может быть и речи.
Причины заболевания
Все белки состоят из аминокислот, которых всего 20, но среди них имеются такие, которые синтезируются в организме человека. Некоторые же должны поступать только извне. К незаменимым аминокислотам относится и фенилаланин. У здорового человека он, попадая внутрь, превращается в тирозин. Это совершенно другая аминокислота, и только несколько процентов вещества направляется в почки и там преобразуется в фенилкетон, достаточно токсичное вещество.
Если у человека отсутствует фермент фенилаланин-4-гидроксилаза или неправильно работает тот, который превращает фенилаланин в иное вещество, то вот в этом случае и развивается фенилкетонурия. Что это достаточно серьезный симптом, скажет вам каждый врач, поэтому необходимо принимать срочные меры.
А привести к отсутствию нужного фермента может мутация гена в хромосоме 12.
Разновидности фенилкетонурии
Если рассматривать формы заболевания, то они могут быть такими:
- Классическая. В этом случае мы наблюдаем, что фенилкетонурия – рецессивный признак. Встречается такая форма у одного ребенка на десять тысяч здоровых детей. Если не принять меры, то едва ли больной человек доживет до тридцати лет.
- Вариативная форма. Не передается по наследству, а вызывает ее мутация в генах. Течение ее более тяжелое, а ранняя смертность прогнозируется с вероятностью практически 100%.

Кроме форм, врачи различают еще и типы фенилкетонурии:
- Первый тип характеризуется тем, что наблюдается недостаток фермента фенилаланин-4-гидроксилазы, который и отвечает за превращение фенилаланина. В 98% случаев диагностируется именно он.
- Второй. Его отличает малое содержание фермента дигидроптеридинредуктазы. У таких больных наблюдаются судороги, а также умственная отсталость. Несмотря на свою редкую встречаемость, смертность от этого типа может наступать в 2-3-летнем возрасте.
- Третий тип характеризуется тем, что возникает дефицит тетрагидробиоптерина. В результате происходит уменьшение объемов мозга, что приводит к умственной отсталости.
Симптоматика болезни
Сразу после рождения ребенка по его внешнему виду или поведению трудно диагностировать заболевание. Основные признаки начнут проявляться немного погодя. Однако еще в родильном доме медики вполне способны поставить диагноз «фенилкетонурия». Симптомы у этого заболевания следующие:
- частая рвота без видимых причин;
- плаксивость;
- вялость;
- могут появляться высыпания по всему телу;
- моча имеет «мышиный» запах;
- ребенок отстает в физическом и умственном развитии от своих сверстников.
Достаточно взять анализ крови и мочи, чтобы поставить правильный диагноз.
Признаки фенилкетонурии
Постепенно при отсутствии надлежащего лечения у больного можно будет наблюдать такие признаки:
- Судорожный синдром. Он начинает проявляться в раннем детстве и сохраняется у взрослых.
- Недостаток пигмента в коже и волосах. Поэтому такие пациенты, как правило, светловолосые и имеют белую кожу.
- Воспалительные процессы, которые по незнанию можно принять за аллергическую реакцию.

Первые признаки умственного отставания можно заметить у ребенка уже в возрасте полугода. Он перестает запоминать новую информацию, и кажется, что он совершенно не способен к обучению. Насторожиться родители должны и тогда, когда малыш забывает то, чему уже давно научился, например, как держать ложку, сидеть, играть с погремушкой. Тревогу нужно бить и в том случае, если ребенок перестает узнавать родителей и близких людей, а также чрезмерная плаксивость с возрастом не проходит.
Вот признаки, которые имеет фенилкетонурия, симптомы болезни необходимо рассматривать только в комплексе, потому что по отдельности они вполне могут встречаться и у здоровых детей.
Выявление заболевания
Поставить правильный диагноз можно двумя путями:
- Провести анализ крови и мочи у новорожденного еще в родильном доме. это, как правило, делают во всех случаях.
- Определить наличие фенилкетонов в биологических жидкостях взрослого человека при наличии соответствующих признаков.
У детей в роддоме кровь берут на 4-5-й день и определяют содержание фенилаланина. Если таковой обнаруживается, то ребенка с мамой направляют на консультацию к генетику.
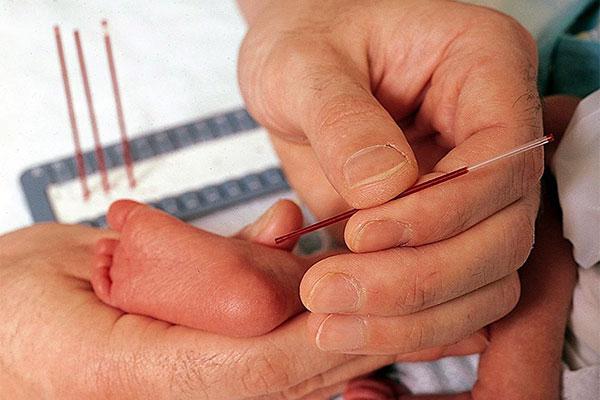
Перед выпиской обязательно поинтересуйтесь, делали ли вашему ребенку анализ на фенилкетонурию. Несмотря на небольшую встречаемость этого заболевания, лучшим решением будет все равно подстраховаться.
Наследование
Так как фенилкетонурия наследуется как рецессивный признак, то для проявления его у ребенка необходимо, чтобы у обоих родителей имелся дефектный ген. Именно поэтому родственные браки во многих странах запрещены.
Если рассматривать случай рождения детей в обычной семье, то у носителей такой мутации они могут быть:
- 25% вероятности, что ребенок родится больным.
- В 50% случаев малыш здоров, но является носителем дефектного гена.
- Четвертая часть потомства будет абсолютно нормальной.
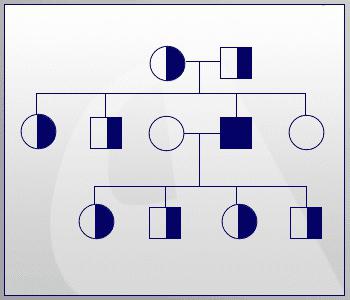
Эта схема не дает полной картины рождаемости больных детей. Она только отражает вероятность, поэтому у каждой семейной пары процент дефектных генов может быть свой, и предсказать исход, к сожалению, невозможно. Сейчас существуют консультации, в которых специалисты-генетики помогают парам спрогнозировать рождение у них больного ребенка, рассказывая при этом, как фенилкетонурия наследуется.
Лечение
Как только ребенку поставлен такой диагноз, меры необходимо принимать незамедлительно. В первую очередь, нужно исключить из рациона белковые продукты. Соблюдать такое строгое ограничение необходимо до 10-12 лет, а лучше и всю жизнь.
Так как младенцы находятся на грудном вскармливании и обычно ничего, кроме материнского молока, еще не употребляют, то врачи рекомендуют маме сократить его потребление ребенком. Сделать это возможно только при одном условии: давать малышу сцеженное молоко, чтобы точно видеть его количество.
Докармливать придется смесями, которые в своем составе не имеют фенилаланина. Когда наступит время введения прикорма, необходимо выбирать пюре и каши без добавления молока. Можно давать соки, овощные пюре.
Врач обязательно назначает и медикаментозное лечение. Обычно это препараты, которые содержат фосфор, ведь этот элемент не зря считают «элементом жизни и мысли», так как он играет важную роль в работе нашего мозга. Также назначаются лекарства, содержащие железо, кальций, они помогают улучшить кровообращение и мозговую деятельность.
Лечение не должно сводиться к полному исключению фенилаланина, так как в этом случае может наблюдаться его недостаток, что приводит к упадку сил, потере аппетита. Кроме того, начинается понос и возникают высыпания на коже.
Чтобы узнать, насколько лечение эффективно, следует периодически сдавать анализ крови и мочи на содержание фенилаланина.
Заболевание у детей
Именно в детском возрасте организм развивается такими темпами, которых не будет в другие периоды жизни. Поэтому в это время важно принять все меры для нормального развития нервной системы. Дети, больные фенилкетонурией, нуждаются не только в приеме медицинских препаратов и специальном питании, но и особом к себе отношении.
Прежде всего, это постоянное внимание, чтобы от зоркого глаза матери не укрылись малейшие отклонения в развитии. Можно применять следующие методы лечения:
- лечебная физкультура, которая поможет ребенку нормально развиваться физически;
- массаж;
- психологическая помощь;
- коррекционная педагогика.

Родители должны понимать, что жизнь и здоровье их ребенка будет в большей степени зависеть от них самих. Какую обстановку они смогут создать вокруг больного малыша, насколько точно будут соблюдены рекомендации врачей по питанию, будут ли близкие люди реагировать на отклонения в умственном и физическом развитии – все эти моменты очень важны.
Народная медицина для избавления от недуга
Народные рецепты находят свое применение при лечении многих болезней. Не исключение и фенилкетонурия. Что это заболевание потребует пересмотра всего образа жизни – это факт. Ребенок должен расти и иметь представление о своем недуге. Родители обязаны в доступной форме объяснить ему, когда он будет способен осознавать полученную информацию, насколько серьезно его положение. Диету и лечение необходимо соблюдать в течение всей жизни. Только в этом случае можно гарантировать полноценное существование.
Народные лекари при фенилкетонурии рекомендуют употреблять больше растительных белков. В такой пище фенилаланина гораздо меньше, чем в животных продуктах. Не запрещается в рацион вводить фрукты и овощи. В них много витаминов и микроэлементов, которые в обязательном порядке необходимы для нормальной работы нервной системы. То есть народная медицина придерживается мнения, что такому больному желательно соблюдать вегетарианскую диету.
Питание при фенилкетонурии
Фенилаланин содержится практически во всех продуктах, в которых есть белок. Необходимо постараться исключить их из своего рациона, и в первую очередь это касается молока и мяса.
Если поставлен диагноз «фенилкетонурия», питание должно быть пересмотрено в первую очередь. Все продукты при этом можно разделить на несколько групп:
- Разрешены к использованию всегда: картофель, сахар, чай, растительные масла.
- Можно употреблять в небольшом количестве: рис, мед, масло сливочное, хлебобулочные изделия, овощи и фрукты.
Полностью необходимо исключить из своего меню: яйца, рыбу и мясо, молоко, макароны, бобовые, орехи, кукурузу, молочные продукты, шоколад.

Учитывая тот факт, что фенилаланин превращается в здоровом организме в тирозин, то больные фенилкетонурией должны в рацион включать продукты, содержащие его в достаточном количестве. К такой пище можно отнести грибы и растительные составляющие.
Прогноз на будущее при фенилкетонурии
Очевидно, что данное заболевание требует немедленного принятия мер, в противном случае жизнь человека будет короткой.
Заболевание «фенилкетонурия» требует внимательного отношения к пациенту. Если соблюдать строгую диету и выполнять все рекомендации врачей, то ребенок сможет нормально расти и развиваться. Прогноз будет также зависеть от того, какие заболевания сопутствуют генетическому недугу и есть ли другие патологии.
Постепенно, с возрастом, организм может в какой-то мере приспосабливаться к повышенному содержанию фенилаланина, поэтому можно иногда допускать послабления в диете. Главное, чтобы не увлечься этими слабостями и вовремя остановиться и перейти на правильное питание.
Если женщина страдает этим недугом, то ей придется еще строже относиться к соблюдению всех рекомендаций, ведь она – будущая мама. Только в этом случае у нее есть возможность родить здорового ребенка.
Это особенно актуально, если учитывать, что методов профилактики данного заболевания практически не существует.
Фенилкетонурия ( Болезнь Феллинга , Фенилпировиноградная олигофрения )
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
МКБ-10
- Причины фенилкетонурии
- Патогенез
- Симптомы фенилкетонурии
- Диагностика
- Лечение фенилкетонурии
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
- Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
- Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
- Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
- Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др. Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.