Анемия фанкони у детей

Впервые столкнувшись с диагнозом «анемия Фанкони», любой человек задастся вполне логичным вопросом – что это такое и как эта патология проявляется. Анемия Фанкони – это генетическая болезнь, которая встречается очень редко. При этом человек страдает от нарушения кровообращения, выражающееся в снижении численности всех клеток крови.
Впервые о заболевании стало известно в 1927 году благодаря швейцарскому педиатру Гвидо Фанкони. Он описал историю болезни трех мальчиков из одной семьи, которые страдали панцитопенией, также у них были обнаружены определенные отклонения в физическом развитии.
Анемия Фанкони диагностируется редко, примерно у пяти человек из 1 миллиона в год. Чаще всего от нее страдают жители Южной Африки. Подавляющее число заболевших являются мальчиками.
Причины
Анемия Фанкони обычно наследуется по аутосомно-рецессивному типу, т.е. вероятность рождения больного ребенка у двух носителей генетической мутации составляет 1/4. В некоторых случаях механизм наследования меняется, и до 1/2 повышается вероятность рождения больного сына у матери-носительницы. Этим объясняется преобладание лиц мужского пола среди больных с верифицированной анемией Фанкони. Выявлено 15 генов, мутации в которых могут приводить к развитию заболевания. Рассматривается гипотеза о причинно-следственной ассоциации болезни с дефектом стволовых клеток.
Причины
Заболевание является наследственным, развивается при передаче дефектного гена от родителей к ребенку. Выявлено 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена находится в X-хромосоме (сцепленной с полом). Все эти гены отвечают за производство определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что и отец, и мать должны быть носителями патологической генетической информации. При этом сами они, как правило, здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, получивших от каждого из родителей один и тот же измененный ген. В крайне редких случаях анемия провоцируется передачей дефектной Х-сцепленной хромосомы. Женщины могут быть носительницами мутации, заболевание проявляется только у мальчиков. Риск развития патологии у сына при наличии у матери мутированного гена – 50%.
Патогенез анемии
У каждой разновидности анемии имеется своя причина, но их патогенез очень схож, поэтому рассмотрим механизм развития часто встречающихся анемий.
Патогенез железодефицитной анемии складывается из двух механизмов:
- Встречается при всех анемиях: гипоксические изменения (нарушение функции тканевого дыхания) в тканях и органах, возникающие в результате снижения количества гемоглобина в организме. Клинические проявления гипоксических изменений представлены общей слабостью, сонливостью, снижением умственной работоспособности и физической выносливости, шумом в ушах, головокружениями, обморочными состояниями, одышкой при нагрузке, сердцебиением, бледностью.
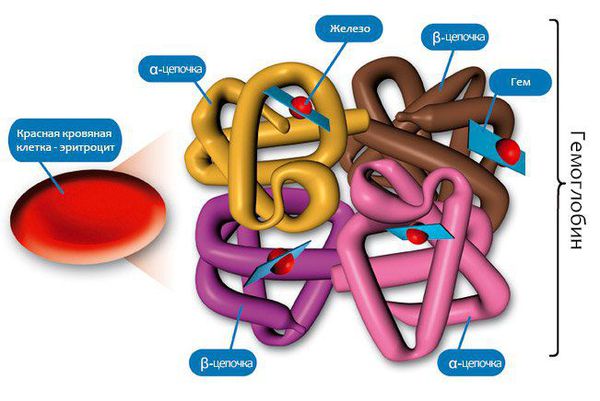
- Присущ исключительно железодефицитной анемии: замедление активности ферментов, содержащих железо, участвующих в тканевом дыхании клеток. Основой формирования железодефицитной анемии является дефицит железа в организме. Железо является каркасом для образования молекул гемоглобина, а именно его железосодержащей части — гема.
Дефицит железа, как и другие анемии в организме, проходит три стадии:
- Прелатентная — скрытая форма, так как эта стадия не имеет клинических проявлений.
- Латентная — нарушается тканевое дыхание и ферментативная активность.
- Анемия — снижение общего количества гемоглобина или гематокрита, уменьшение количества эритроцитов [1] .
Патогенез В12-дефицитной анемии
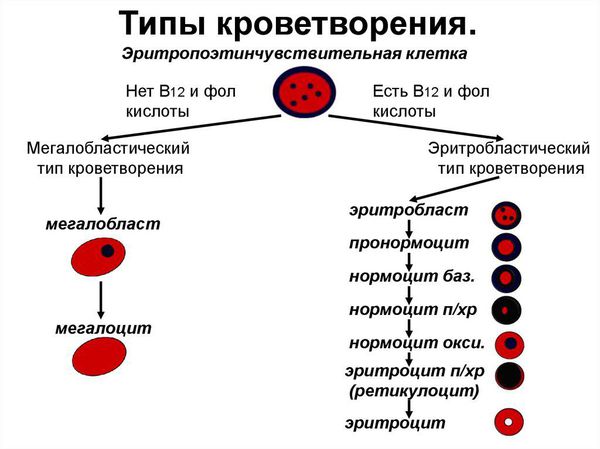
Сущность В12 дефицитной анемии заключается в нарушении процесса созревания красных кровяных клеток вследствие дефицита витамина В12 или В9 (фолиевой кислоты).
При дефиците витамина В12 прежде всего страдают: костный мозг, головной мозг и эпителий желудочно-кишечного тракта. В красном ростке кроветворения появляется мегалобластный тип кроветворения: возникает дефект синтеза ДНК, в результате чего формируются очень крупные клетки — мегалобласты, а из них мегалоциты; нарушается синхронность созревания ядра и цитоплазмы эритроцитов; красные кроветворные клетки гибнут рано. В это же время снижается гранулоцитопоэз (образование гранулоцитов, которые представляют собой самую многочисленную группу лейкоцитов) и тромбоцитопоэз (образование тромбоцитов).
При недостатке витамина В12 нарушается обмен жирных кислот и накапливаются нейротоксичные метилмалоновая и пропионовая кислоты. Синтез миелина (вещества, образующего миелиновую оболочку нервных волокон) нарушается. Повреждаются задние и боковые спинномозговые столбы [3] .
Диагностика

Для подтверждения диагноза необходима консультация гематолога. С учетом причин анемии привлекают других специалистов — гастроэнтеролога, инфекциониста, кардиолога, пульмонолога, гинеколога, ревматолога, нефролога, дерматолога, хирурга.
Основу диагностики составляет внешний осмотр и лабораторные исследования.
Врач оценивает состояние и цвет кожи, ногтей и волос, пальпаторно определяет размер селезенки и печени. Обращает внимание на пульс, сердечный ритм, показатели артериального давления, характер и частоту дыхания.
Наиболее информативные методы исследования:
- Общий анализ крови. При малокровии легкой степени уровень гемоглобина составляет 110‒90 г/л, количество эритроцитов до 3,5х1012/л; средней степени — гемоглобин 90‒70 г/л, эритроциты до 2,5х1012/л; тяжелой степени — гемоглобин 70 г/л и ниже, эритроциты менее 2,5х1012/л.
- Анализ на ферритин.Ферритин — основной индикатор запаса железа. Исследование позволяет диагностировать избыток или дефицит гемоглобина, дифференцировать вид анемии, заподозрить опухоли, воспалительные и инфекционные заболевания.
- Анализ на трансферрин. Это сывороточный белок, который отвечает за перенос железа. На основании результатов определяют различные формы малокровия. На показатели уровня трансферрина влияет функциональное состояние печени, нарушения всасываемости веществ в кишечнике.
Для подтверждения некоторых форм анемии достаточно только общего анализа крови. Дифференциальную диагностику проводят между разными формами анемии. Для установления причин дефицита железа назначают дополнительные инструментальные и лабораторные исследования.
Лабораторные признаки анемии Фанкони
Трёхростковая аплазия выступает наиболее типичной манифестацией анемии Фанкони, однако наблюдения за инициально гематологически интактными гомозиготами показали, что зачастую тромбоцито- или лейкопения предшествуют развитию панцитопении. Первые гематологические аномалии при анемии Фанкони закономерно обнаруживают после респираторных вирусных инфекций, прививок, иногда гепатитов — так, как это характерно и для идиопатических апластических анемий. Для анемии Фанкони даже в доанемическую фазу типичен выраженный макроцитоз, сопровождающийся значительным повышением уровня фетального гемоглобина. Пунктат костного мозга, как правило, обеднён кроветворными клеточными элементами, преобладают лимфоциты, встречаются плазматические, тучные клетки и стромальные элементы — клиническая картина, неотличимая от идиопатической апластической анемии. Зачастую в аспирате костного мозга обнаруживают дисмиелопоэз и дизэритропоэз, в частности, мегалобластоидность, благодаря которой Фанкони назвал эту анемию «пернициозиформной». В биоптатах костного мозга на ранних стадиях заболевания выявляются гиперклеточные участки активного резидуального гемопоэза, которые исчезают по мере прогрессирования заболевания.
Один из фундаментальных феноменов, характерных для клеток крови больных анемией Фанкони, — это их склонность к формированию специфических хромосомных аномалий — разрывов, сестринских обменов, эндоредупликаций при культивировании клеток in vitro. Инкубация ФГА-стимулированных лимфоцитов больных анемией Фанкони с бифункциональными алкилирующими агентами, которые вызывают сшивки ДНК между гуанидиновыми основаниями, расположенными как на одной, так и на двух комплементарных цепях — нитроген-мустардом, препаратами платины, митомицином и особенно диэпоксибутаном — резко увеличивает количество аберраций. Этот феномен, получивший название кластогенного эффекта, лежит в основе современной диагностики и дифференциальной диагностики анемии Фанкони, поскольку спонтанные аберрации могут как отсутствовать у больных анемией Фанкони, так и присутствовать у больных с другими синдромами, в частности с синдромом Ниймеген. Под влиянием бифункциональных алкилирующих агентов происходит замедление клеточного цикла: клетки больных анемией Фанкони останавливаются в G2 фазе митотического цикла, что послужило основанием для разработки ещё одного диагностического теста для анемии Фанкони с помощью метода проточной флюориметрии.
Возраст первого появления анемии Фанкони в одной семье часто конкордантен, но может и существенно варьировать, в том числе и у однояйцевых близнецов. В прошлом при отсутствии специфического лечения (андрогены или трансплантация костного мозга) и проведении только гемотрансфузий заболевание неуклонно прогрессировало: 80% больных умирали от осложнений панцитопении в течение 2 лет после установления диагноза апластической анемии и практически все больные умирали через 4 года. Необходимо упомянуть, что зафиксировано несколько случаев спонтанного улучшения и даже полного восстановления гематологических показателей.
Вторыми по частоте развития гематологической презентацией анемии Фанкони выступают острые лейкозы и миелодиспластические синдромы. Примерно у 10% больных анемией Фанкони, клинические случаи которых описаны в литературе, впоследствии развился острый лейкоз. Во всех случаях, за исключением 2, лейкозы были миелоидными. Описаны даже случаи установления диагноза анемии Фанкони у пациента с резидуальной цитопенией через много лет после успешной химиотерапии ОМЛ. Несколько ниже частота развития миелодиспластических синдромов — около 5%, причём только у 1/5 из этих больных прослежена дальнейшая эволюция МДС в ОМЛ и несколько больных с МДС прожили более 10 лет. Согласно исследованиям Международного регистра анемии Фанкони риск развития ОМЛ или МДС у больных анемией Фанкони равен 52% к 40 годам. Зачастую выявляют кариотипические аномалии (моносомию 7, трисомию 21, делецию 1), которые позволяют квалифицировать ОМЛ и МДС у больных анемией Фанкони как вторичные. Интересно, что, хотя риск развития МДС/ОМЛ у больных с хромосомными аномалиями примерно в 10 раз выше, чем без таковых, наличие хромосомных аберраций не означает обязательного развития МДС. Клоны, несущие аномалии, могут спонтанно исчезать или сменять друг друга.
Кроме гематологических аномалий для больных анемией Фанкони характерна склонность к развитию опухолей. Риск развития злокачественных опухолей у больных анемией Фанкони составляет 10%, из них 5% приходится на долю опухолей печени и 5% — на остальные опухоли. Опухоли реже встречаются у детей — средний возраст диагностики опухолей печени составляет 16 лет, а остальных опухолей — 23 года. Опухоли печени (гепатоцеллюлярная карцинома, гепатома, аденома и др.), а также пелиоз («кровяные озерца») встречаются чаще у мужчин (соотношение 1,6:1), причём применение андрогенов увеличивает риск их возникновения. В то же время внепечёночные опухоли чаще встречаются у женщин (соотношение 3:1) даже после исключения опухолей гинекологической сферы. Самые частые формы рака при анемии Фанкони — чешуйчатоклеточные карциномы языка и рак пищевода, на которые приходится более 30% всех внепечёночных опухолей при анемии Фанкони, остальные опухоли встречаются в 5-7 раз реже.