Синдром Гарднера
Синдром Гарднера – это наследственное заболевание, сопровождающееся полипозом толстого кишечника в сочетании с доброкачественными неоплазиями кожи, костей и мягких тканей. Может долгое время протекать бессимптомно. Возможны вздутие живота, урчание и расстройства стула. В некоторых случаях полипоз кишечника осложняется кровотечением или кишечной непроходимостью. Отмечается высокая вероятность развития колоректального рака. Заболевание диагностируется на основании жалоб, семейного анамнеза, данных осмотра, рентгенографии, КТ, МРТ, УЗИ, эндоскопии и других исследований. Лечение – эндоскопическая полипэктомия или резекция пораженных отделов кишечника.
МКБ-10
- Причины
- Симптомы
- Диагностика
- Лечение синдрома Гарднера
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Синдром Гарднера – редкая генетически обусловленная патология, при которой наблюдается диффузный полипоз толстого кишечника в сочетании с доброкачественными опухолями костей и мягких тканей (остеомами, фибромами, нейрофибромами, эпителиальными кистами и другими неоплазиями). Полипозом при синдроме Гарднера преимущественно поражаются прямая и сигмовидная кишка, однако полипы могут выявляться в других отделах кишечника. Впервые был описан американским врачом и генетиком Е. Дж. Гарднером в 1951 году. С тех пор в специальной литературе появились упоминания более чем о ста случаях данного заболевания. Риск малигнизации полипов толстой кишки с развитием колоректального рака в течение жизни составляет около 95%. Лечение проводят специалисты в сфере клинической проктологии, гастроэнтерологии, онкологии, ортопедии, стоматологии и челюстно-лицевой хирургии.
Причины
Синдром Гарднера передается по аутосомно-доминантному типу. Выраженность кишечных и внекишечных клинических проявлений может сильно варьировать. Первые симптомы синдрома Гарднера обычно появляются у детей старше 10 лет. Возможно позднее начало с образованием первых опухолей в возрасте старше 20 лет. В отдельных случаях наряду с полипозом толстого кишечника, остеомами и мягкотканными новообразованиями у больных синдромом Гарднера обнаруживаются полипы тонкого кишечника, желудка и двенадцатиперстной кишки.
Симптомы
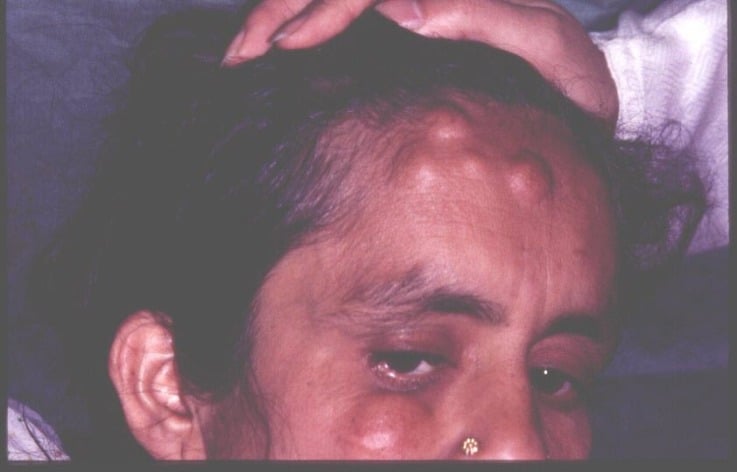
Синдром Гарднера включает в себя характерную триаду: диффузный полипоз нижних отделов толстого кишечника, остеомы плоских и трубчатых костей, различные доброкачественные опухоли кожи и мягких тканей. При умеренном количестве и небольшом размере полипов кишечные проявления синдрома Гарднера могут отсутствовать или быть слабо выраженными. В подростковом или юношеском возрасте больные обычно впервые обращаются к врачам в связи с появлением доброкачественных костных и мягкотканных опухолей.
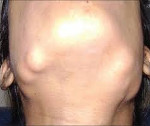
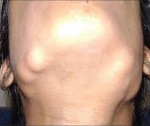
Остеомы при синдроме Гарднера могут локализоваться как в плоских, так и в трубчатых костях. Часто наблюдается поражение костей лицевого черепа, сопровождающееся обезображиванием. Возможно смещение и даже выпадение зубов. Через некоторое время после появления рост остеом у больных синдромом Гарднера прекращается, опухоли не озлокачествляются. Неоплазии мягких тканей отличаются большим разнообразием. Особенно часто выявляются липомы, дерматофибромы, нейрофибромы и эпителиальные кисты. Реже встречаются атеромы, лейомиомы и другие новообразования. Мягкотканные опухоли при синдроме Гарднера также протекают доброкачественно, малигнизация отсутствует.
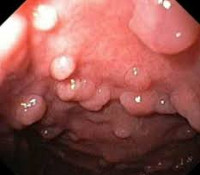
Полипы толстой кишки при синдроме Гарднера нередко становятся случайной находкой при проведении исследований ЖКТ по другим поводам либо обнаруживаются в процессе расширенного обследования, назначенного в связи с появлением множественных мягкотканных и костных неоплазий. В течении синдрома Гарднера можно выделить три стадии поражения кишечника. На первой стадии заболевание протекает бессимптомно. На второй пациенты отмечают дискомфорт в животе, вздутие, урчание и периодические нарушения стула. В каловых массах могут обнаруживаться примеси крови и слизи.
На третьей стадии у больных синдромом Гарднера выявляются выраженный болевой синдром, постоянный метеоризм, обильные примеси слизи и крови в испражнениях, снижение веса, повышенная утомляемость, эмоциональная лабильность, нарушения электролитного и белкового обмена. У многих пациентов с синдромом Гарднера развивается анемия, обусловленная небольшими по объему, но часто повторяющимися кровотечениями из нижних отделов ЖКТ. В отдельных случаях у больных развиваются неотложные состояния, требующие экстренной медицинской помощи – обильные кишечные кровотечения или кишечная непроходимость.
Диагностика
Диагноз устанавливается на основании семейного анамнеза (наличия синдрома Гарднера у близких родственников), клинической картины, включающей в себя характерную триаду, и данных дополнительных исследований. При проведении физикального осмотра врач отмечает наличие множественных костных и мягкотканных опухолей различной локализации. У некоторых больных синдромом Гарднера выявляются деформации лица, обусловленные остеомами лицевого черепа. При пальпации костей туловища и конечностей могут обнаруживаться опухолевидные образования костной плотности. При поражениях легкой степени количество неоплазий может быть незначительным, что затрудняет диагностику.
При пальпации живота наблюдается болезненность в левой подвздошной области. На первой стадии поражения кишечника данный симптом может отсутствовать. При проведении пальцевого ректального исследования на слизистой прямой кишки больных синдромом Гарднера обнаруживаются множественные узлы. На контрастных рентгеновских снимках такие узлы отображаются в виде дефектов наполнения. При узлах небольшого размера (менее 1 см) информативность контрастного рентгенологического исследования снижается. В ходе ректороманоскопии выявляются полипы в прямой и ободочной кишке. Количество полипов может сильно варьировать.
У некоторых пациентов с синдромом Гарднера отмечаются ограниченные поражения отдельных участков кишки. В отличие от рентгенографии, эндоскопическое исследование дает возможность диагностировать полипы любого размера, в том числе – мелкие (диаметром от 1-2 мм). Для уточнения характера и распространенности костных опухолей при синдроме Гарднера осуществляют рентгенографию. При мягкотканных новообразованиях назначают КТ, МРТ или УЗИ области поражения. При необходимости выполняют биопсию полипов, остеом и мягкотканных новообразований.
Дифференциальную диагностику синдрома Гарднера врачи-проктологи проводят с обычными множественными полипами и другими формами семейного полипоза. Для разных вариантов наследственного полипоза характерны определенные отличия в преимущественной локализации полипов (поражение всего толстого кишечника, поражение дистальных отделов толстой кишки), характере патологических изменений костей и мягких тканей. Для уточнения этих различий перед постановкой окончательного диагноза проводят детальный внешний осмотр, осуществляют ирригоскопию и колоноскопию.
Лечение синдрома Гарднера
Лечение только хирургическое. Поскольку риск озлокачествления костных и мягкотканных неоплазий отсутствует, решение о проведении оперативных вмешательств принимают при наличии косметического или функционального дефекта. Полипоз толстого кишечника при синдроме Гарднера рассматривается, как облигатный предрак, поэтому многие врачи считают целесообразным проведение операции до появления признаков малигнизации. При небольшом количестве полипов возможна эндоскопическая полипэктомия.
При синдроме Гарднера с выраженным диффузным полипозом показана резекция пораженного участка кишечника или тотальная колэктомия с наложением илеостомы либо формированием илеоректального анастомоза (при отсутствии полипов прямой кишки). Хирургическое вмешательство рекомендуют проводить в возрасте 20-25 лет. Из-за калечащего характера операции молодые пациенты с синдромом Гарднера нередко отказываются от данного вмешательства. В подобных случаях показано динамическое наблюдение с проведением колоноскопии через каждые 6-8 месяцев.
Некоторые врачи являются сторонниками выжидательной тактики и считают, что колэктомию при синдроме Гарднера следует проводить только при появлении признаков озлокачествления или при часто повторяющихся кровотечениях с развитием анемии. Показанием к экстренному оперативному вмешательству при синдроме Гарднера являются обильное кишечное кровотечение и кишечная непроходимость.
Прогноз и профилактика
При своевременном адекватном лечении прогноз при синдроме Гарднера достаточно благоприятный. Тяжесть течения определяется выраженностью полипоза и локализацией внекишечных опухолей. Родителям, имеющим родственников, страдающих данным заболеванием, в период планирования беременности рекомендуют обратиться за медико-генетической консультацией.
Синдром Гарднера
Что такое синдром Гарднера?
Синдром Гарднера представляет собой редкий вариант семейного аденоматозного полипоза — состояние, характеризующееся множественными доброкачественными опухолями в толстой кишке и прямой кишке, которые в конечном итоге могут перерасти в колоректальный рак.
Термин «Гарднер» относится к ученому Элдону Дж. Гарднеру, который впервые описал синдром в 1951 году. Болезнь возникает из-за мутаций в гене Adenomatous Polyposis Coli (APC) — гене-супрессоре опухоли, который кодирует белки, ответственные за контроль деление и рост клеток. Мутация в этом гене запускает неконтролируемый рост клеток, что впоследствии приводит к образованию полипов или опухолей.
Синдром Гарднера может привести к изменениям в различных частях тела. Опухоли чаще всего появляются в толстой кишке, иногда в большом количестве. С возрастом они, как правило, увеличиваются. Помимо полипов в толстой кишке, синдром может вызывать появление фиброидов, десмоидных опухолей, сальных кист, сидячих полипов, гиперпластических полипов, кист нижней челюсти, брыжеечных кист. У некоторых пациентов с синдромом Гарднера могут встречаться даже поражения сетчатки глаза.
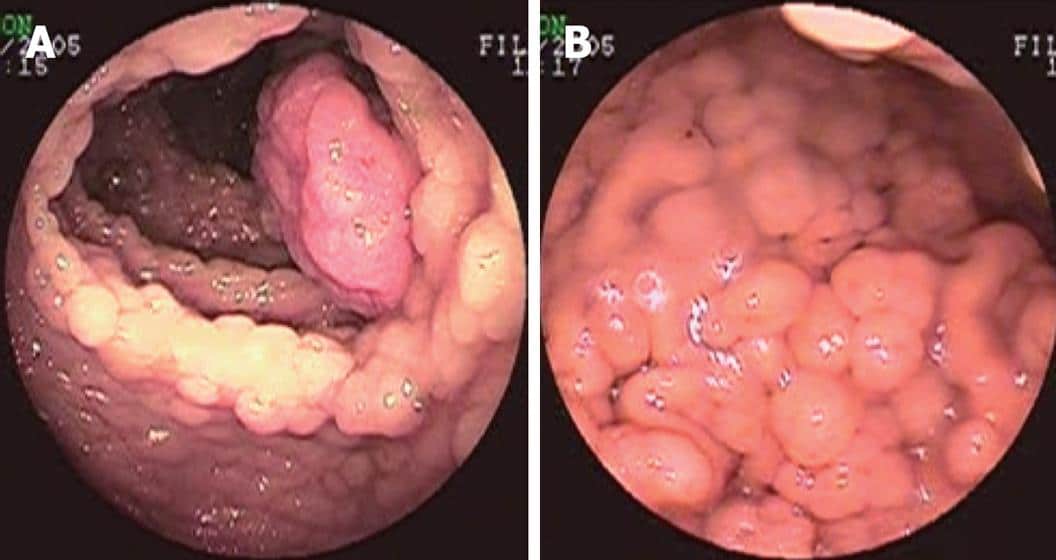
При синдроме Гарднера колоректальный рак (злокачественное новообразование толстого кишечника) развивается примерно в возрасте:
- 39 лет для людей, страдающих классическим семейным аденоматозным полипосом.
- 55 лет для людей, страдающих так называемым семейным аттенуированным аденоматозным полипосом.
Основным фактором риска развития синдрома Гарднера является наличие хотя бы одного родителя с этими заболеваниями. Спонтанная мутация в генах APC встречается гораздо реже.
Частота семейного аденоматозного полипоза, являющегося причиной синдрома Гарднера, колеблется от 1 на 7 000 до 1 на 22 000 человек.
Причины возникновения
Синдром Гарднера — это генетическое заболевание, а значит, наследственное. Ген Adenomatous Polyposis Coli (APC) является медиатором производства белков APC. Белок APC регулирует рост клеток, предотвращая их слишком быстрое или беспорядочное размножение.
У людей с синдромом Гарднера есть дефект гена APC. Это приводит к аномальному росту тканей. Что вызывает мутацию этого гена, пока не установлено.
Классификация синдрома Гарднера
Синдром Гарднера можно классифицировать следующим образом:
- классический тип семейного аденоматозного полипоза;
- семейный аттенуированный аденоматозный полипоз;
- семейный аденоматозный полипоз аутосомно-рецессивного типа.
У людей с классическим типом количество полипов увеличивается с возрастом до сотен тысяч, а семейный аттенуированный аденоматозный полипоз представляет собой, наоборот, вариацию развития расстройств, при которых происходит задержка роста полипа.
Люди с аутосомно-рецессивным типом заболевания страдают от более мягкой формы этого заболевания и имеют меньше полипов, чем люди с классическим типом. В последнем варианте развивается менее 100 полипов, что обусловлено мутациями в генах, отличительных от классического и семейного аттенуированного аденоматозного полипоза.
Особое значение имеют также нераковые образования, называемые десмоидными опухолями. Эти волокнистые опухоли обычно возникают в тканях, покрывающих кишечник, и могут быть спровоцированы операцией по удалению толстой кишки. Десмоидные опухоли имеют тенденцию повторяться и после хирургического удаления.
В каких ещё частях тела развивается синдром?
При синдроме Гарднера как при классическом, так и при аттенуированном полипозе доброкачественные и злокачественные опухоли могут развиваться в других частях тела, таких как:
Симптомы и признаки
Симптомы синдрома Гарднера у разных пациентов отличаются. Главной особенностью этого расстройства является наличие полипов толстой кишки, что проявляется у 80% — 99% пациентов.
Другие симптомы, связанные с синдромом Гарднера, включают в себя:
- полипы в желудке и тонкой кишке;
- зубные аномалии;
- доброкачественные новообразования костей или остеомы;
- доброкачественные опухоли кожи и соединительной ткани, называемые эпидермоидными кистами и десмоидными опухолями;
- пигментная эпителиальная гипертрофия сетчатки.
Диагностика
Несколько молекулярно-генетических тестов, которые обнаруживают мутацию в гене APC, доступны для диагностики синдрома Гарднера. Эти тесты включают генотипирование, исследование секвенирования, анализ числа копий генов и эпигенетическое тестирование.
Если семейный анамнез заболевания известен, анализ последовательности носителей является хорошим вариантом для будущих родителей, чтобы выяснить, являются ли они носителями вызывающей болезнь мутации. Это жизненно важный шаг на пути предотвращения передачи болезней следующему поколению.
Помимо генетического тестирования, проводиться рутинная диагностика синдрома Гарднера — это колоноскопия и рентгенологическое исследование длинных костей и челюстных костей. Обнаружение дополнительных признаков, таких как пигментные поражения в сетчатке, кожные аномалии и полипы в желудке и тонкой кишке, также подтверждает диагноз синдрома Гарднера.
Лечение синдрома Гарднера
Поскольку доброкачественные полипы в толстой кишке и прямой кишке появляются в среднем в возрасте 16 лет, регулярный скрининг пациентов начинается уже в возрасте 10 лет. Если симптомы не лечить, эти полипы могут превратиться в злокачественные опухоли в среднем в возрасте 40 лет.
Следовательно, ежегодный скрининг является лучшим способом предотвращения неблагоприятных последствий этого заболевания. Ежегодные осмотры с использованием колоноскопии, эзофагогастродуоденоскопии и обычных физических осмотров являются основной схемой лечения заболеваний.
Хирургическое удаление толстой кишки рекомендуется у пациентов с множественными запущенными полипами. Этим пациентам обычно назначают нестероидные противовоспалительные препараты (НПВП) для лечения оставшихся полипов.
Для десмоидных опухолей рекомендуется хирургическое удаление или лечение антиэстрогенами, НПВП, химиотерапия и радиотерапия. Эпидермоидные кисты обычно лечат так же, как обычные кисты, и иногда требуются удаления или инъекции стероидов.
Если присутствуют зубные аномалии, для устранения этих проблем может быть рекомендовано ортодонтическое лечение.
Как и при любых других заболеваниях, здоровый образ жизни с адекватным питанием, физическими упражнениями и снятием стресса может помочь людям справиться с физическими и эмоциональными проблемами.
Фармакотерапия
Такие препараты, как нестероидные противовоспалительные препараты (НПВП), могут помочь предотвратить прогрессирование колоректальных полипов до карциномы при семейном аденоматозном полипозе, одним из которых является синдром Гарднера.
Было показано, что они уменьшают размер и количество полипов в течение всего периода введения, но это не является неизменным, поскольку у некоторых пациентов одновременно появляются новые опухоли или злокачественная трансформация существующих полипов.
Более новые препараты в этой категории включают применение ингибиторов ЦОГ-2 поскольку рецепторы ЦОГ-2 присутствуют в слизистой оболочке толстой кишки, подвергаясь раковым изменениям. Результаты клинических испытаний показали, что размер полипов уменьшился, но влияние на уровень злокачественности не было определено.
Синдром Гарднера и рак
Кишечные полипы являются основной проблемой для пациентов с синдромом Гарднера из-за их склонности к раку. Однако полное удаление толстой кишки и большей части прямой кишки увеличивает 5-летнюю выживаемость пациентов почти до 100%.
Симптомы, такие как полипы кожи или остеомы, в первую очередь представляют косметическую проблему и должны лечиться, чтобы уменьшить видимый дискомфорт.
Какова перспектива жизни людей, затронутых этой болезнью?
Перспективы (прогноз) для людей с синдромом Гарднера варьируются, в зависимости от тяжести симптомов. Люди, у которых есть мутация гена APC, например, страдающие синдромом Гарднера, с возрастом все чаще заболевают раком толстой кишки.
Без хирургического лечения почти у всех людей с мутацией гена APC развивается рак толстой кишки к 39 годам (в среднем).
Синдром Гарднера: симптомы, профилактика
Синдром Гарднера является аутосомно-доминантным синдромом аденоматозного полипоза толстой кишки, который связан с остеомами и поражениями кожи. Остеомы часто предшествуют любым другим симптомам, в том числе обусловленным полипозом толстой кишки, и, таким образом, могут служить маркером последних.

Синдром Гарднера классифицируется как «абсолютно предраковое» заболевание с риском развития рака от 70 до 90 %.
Ранняя оценка синдрома Гарднера
Ассоциация полипов толстой кишки с раком толстой кишки почти у 100% пациентов с синдромом Гарднера делает важным скрининг пациентов с остеомами на предмет синдрома и, таким образом, потенциально спасает их жизни. Рассмотрение этого диагноза означает, что слизистую оболочку желудка, щитовидную железу, эпителий сетчатки, череп и зубы, а также кожу необходимо оценивать на предмет эпидермоидных кист, десмоидальных опухолей, врожденной гипертрофии пигментного эпителия сетчатки и т. д. Классическим местом образования остеомы является нижняя челюсть, особенно под углом челюсти, но эти опухоли могут также образовываться на черепе, околоносовых пазухах и длинных костях. КТ может помочь показать их местоположение и размер. Чаще всего обнаруживается, что полипы покрывают всю поверхность толстой кишки, но иногда могут возникать в слизистой оболочке желудка и тонкой кишки. Обычно они впервые появляются после полового созревания, но первые симптомы проявляются чаще всего к 40 годам.
Обнаружение других опухолей
Скрининг папиллярного рака щитовидной железы, аденом и аденокарцином надпочечников, гепатоцеллюлярного рака, остеосаркомы и хондросаркомы, а также других опухолей щитовидной железы и печени также является важной частью первоначальной оценки.
Профилактика рака толстой кишки
Рекомендована восстановительная проктоколэктомия с анальным анастомозом подвздошной кишки наряду с мукозэктомией, потому что вся слизистая оболочка толстой кишки резецируется для предотвращения образования полипов, сохраняя при этом функцию кишечника и обеспечивая сексуальное удовлетворение. Избегая необходимости в колостоме, пациент может избежать многих психологических и физических расстройств. Временно вводится илеостомия для правильного заживления ректально-ободочной кишки.
Когда полипы желудка обнаруживаются в связи с синдромом Гарднера, частота и скорость канцерогенеза в таких случаях ниже, чем при колоректальном полипозе. Таким образом, более консервативный подход, такой как малый полипэктомия, может дать хорошие результаты.
Фармакотерапия и профилактика
Такие лекарства, как нестероидные противовоспалительные препараты (НПВП) могут помочь в предотвращении прогрессирования колоректальных полипов в карциному при семейном аденоматозном полипозе, одним из которых является синдром Гарднера. Было показано, что они уменьшают размер и количество полипов в течение всего периода введения, но это не является неизменным, поскольку у некоторых пациентов одновременно появляются новые опухоли или злокачественная трансформация существующих полипов.
Более новые препараты в этой категории включают ингибиторы ЦОГ-2 из-за обнаружения, что рецепторы ЦОГ-2 присутствуют в слизистой оболочке толстой кишки, подвергающейся раковым изменениям, но не в нормальной слизистой оболочке толстой кишки. Результаты клинических испытаний показали, что размер полипов уменьшился, но влияние на уровень злокачественности не было определено.
Наблюдение
Все пациенты с синдромом Гарднера должны регулярно проходить мониторинг тонкой и толстой кишки, печени и щитовидной железы. Также ежегодно должно проводиться обследование щитовидной железы и УЗИ.
Фото превью: glcgb.ru
Встройте «Правду.Ру» в свой информационный поток, если хотите получать оперативные комментарии и новости:
Подпишитесь на наш канал в Яндекс.Дзен или в Яндекс.Чат
Добавьте «Правду.Ру» в свои источники в Яндекс.Новости или News.Google
Также будем рады вам в наших сообществах во ВКонтакте, Фейсбуке, Твиттере, Одноклассниках.
14 самых редких болезней в мире
О самых редких болезнях, причинах их появления, симптомах и жизни с ними вы узнаете в материале uznayvse.ru.
Внимание, некоторые фотографии в материале могут вызвать у вас неприятные чувства.
Моргеллонова болезнь
Большинство пострадавших заявляло о неких «волокнах» или червях, проникших вглубь эпидермиса. Эти «нити» можно увидеть под микроскопом. Многие ученые полагают, что Моргеллон возбуждает неизвестная науке мутация грибка, способного выживать даже при абсолютном нуле.
Версия о психогенной природе заболевания тоже до сих пор популярна. В 2012 году было опубликовано исследование о том, что никаких известных науке болезнетворных организмов у пациентов не найдено, а широкое освещение болезни Моргеллона в СМИ спровоцировало резкий всплеск болезни.
К 2017 году было зарегистрировано около 20 тысяч жалоб с похожими симптомами. География болезни: США (все 50 штатов), реже встречается в Нидерландах, Австралии, Великобритании
Временная слепота
Паранеопластическая пузырчатка
Есть несколько видов пузырчатки – дерматологического заболевания с аутоиммунной природой (Аутоиммунные заболевания – класс болезней, при которых лимфоциты начинают атаковать свои же собственные, здоровые клетки организма). Ее паранеопластическая разновидность встречается реже всего, но очень опасна.
Иммунная система начинает нападать на кератиноциты, составляющие основную массу эпидермиса, из-за чего в нем образуются пустоты, заполняющиеся жидкостью. На этих местах образуются мокрые пузыри, сквозь которые легко проникают внешние инфекции.
Аллергия на воду
Триметиламинурия, или синдром рыбного запаха
Анальгия
Врожденная нечувствительность к боли появляется из-за мутации гена SCN9A и довольно часто встречается у детей до 2-х лет. Но есть случаи – несколько сотен на все население земного шара – когда невосприимчивость сохраняется и во взрослом возрасте. У больных анальгией значительно выше вероятность получения ожога, перелома или сепсиса.
Стивен Пит из Вашингтона, как и его брат-близнец, не понаслышке знает о коварстве этого синдрома. «Мне было 6 лет, я катался на роликах, упал и услышал крик мамы. Смотрю – у меня кость из ноги торчит. Ничего не почувствовал», – вспоминал он. В детстве он ломал левую ногу едва ли не каждый месяц, пока органы опеки не изъяли детей из семьи, заподозрив насильственные действия. Родителям пришлось потратить много времени, сил и красноречия, чтобы доказать свою невиновность.
Болезнь Куру
Аргироз
Аллергия на электричество
Фатальная семейная бессонница
В конце 90-х годов ученым удалось выяснить природу болезни: из-за мутации в 20-й хромосоме аспаргин меняется на аспаргиновую кислоту, в результате белковая молекула трансформировалась в прион. По цепной реакции прион превращает остальные белковые молекулы в себе подобные. В отделе мозга, который отвечает за сон, накапливаются бляшки, которые и вызывают хроническую бессонницу, истощение и смерть.
Выделяется 4 фазы болезни: во время первой человек становится одержим параноидальными идеями, к четвертой перестает реагировать на внешние раздражители. Болезнь длится от 7 до 36 месяцев; лечения нет, не помогают даже сильнейшие снотворные. Всего известно 40 семей, в которых наследуется это заболевание.
Прогрессирующая фибродисплазия
Костная ткань при прогрессирующей фибродисплазии неконтролируемо разрастается за счет соседних мышечных тканей. Запускает патологический процесс чаще всего травма, даже самая небольшая. Причем оперативное вмешательство — не выход. Если вырезать окостеневший участок, это спровоцирует новый очаг роста кости.
Фибродисплазия – это генетическое заболевание, которое передается по наследству и до недавнего времени совсем не поддавалось лечению. В 2006 году группа исследователей Пенсильванского университета открыла ген, отвечающий за эту мутацию. С тех пор были начаты работы над генными блокаторами в гене ACVR1/ALK2.
Детская прогерия
Самая редкая болезнь в мире – болезнь Филдс
В 9 лет Катрин и Кирсти пересели на инвалидные коляски, а в 14 синхронно лишились речи. В 2012 году им выдали речевые устройства, аналогичные тому, которым пользовался Стивен Хокинг. «Теперь нас могут различать по акценту. Я выбрала австралийский, а моя сестра американский. Электронный голос позволяет нам даже спорить друг с другом», – шутила Катрин.
Синдром Гарднера: что означает диагноз и как проявляется, лечение одной из самых редких болезней в мире
Агаммоглобулинемия Х-сцепленная инфантильная (син.: агаммаглобулинемия Брутона, врожденная агаммаглобулинемия). Поражает исключительно мужчин. Тип наследования Х-сцепленный рецессивный. Ген локализован на Х-хромосоме, в области q21.3-q22. В основе заболевания лежат отсутствие В-лимфоцитов, отсутствие или резкое снижение содержания основных классов сывороточных иммуноглобулинов.
Минимальные диагностические признаки агаммоглобулинемии Х-сцепленной инфантильной: рецидивирующие тяжелые бактериальные инфекции, вызванные неспособностью продуцировать функциональные антитела. Поражаются дыхательные пути, желудочно-кишечный тракт и кожа.
Клинически агаммоглобулинемия Х-сцепленная инфантильная характеризуется тяжело протекающими воспалительными процессами, чаще всего отитами, конъюнктивитами, синуситами, энтеритами, рецидивирующими инфекциями верхних дыхательных путей, бронхитами, пневмониями, пиодермиями, вызванными главным образом стафилококками, пневмококками, стрептококками. Очень тяжело протекает гепатит, который может привести к смерти. Возможны полиартрит и дерматомиозит. Нередко состояние осложняется сепсисом.
Больные агаммоглобулинемией Х-сцепленной инфантильной бледны, малоподвижны, на коже лица, туловища, конечностей — очаги пиодермии. В периферической крови обнаруживаются анемия, лейкопения, нейтропения, транзиторная эозинофилия. В-лимфоциты в крови, лимфоидной ткани, костном мозге отсутствуют или их количество резко снижено, как и количество плазматических клеток. Резко снижен уровень иммуноглобулинов: IgM и TgA отсутствуют, уровень IgG. при рождении нормальный, к 6 мес значительно снижается.
Иммунизация не приводит к положительным результатам, изогемагглютинины отсутствуют. При агаммоглобулинемия Х-сцепленной инфантильной отмечается высокая летальность в раннем возрасте. У 5% пациентов в более позднем возрасте развиваются злокачественные лимфопролиферативные заболевания: лейкозы или лимфомы.
Дифференциальный диагноз проводится с агранулоцитозом Кастелмана, вторичными иммунодефицитными состояниями.
Тяжелые комбинированные иммунодефициты (ТКИД) описываются в группе заболеваний, сопровождающихся дефектами гуморального и клеточного иммунитета. Они характеризуются аутосомно-рецессивным или Х-связанным рецессивным типом наследования.
Агаммоглобулинемия Х-сцепленная инфантильная проявляется неукротимым поносом, пневмонией и различными инфекциями, в первую очередь кандидозом. Пиодермические высыпания часто развиваются в течение первых нескольких месяцев жизни, после их заживления остаются гиперпигментированные участки. Почти у 5% больных имеются злокачественные лимфопролиферативные заболе вания.

Кожные заболевания ассоциированные с полипозом кишечника
Ряд наследственных кожных синдромов ассоциируется с опухолями желудочно-кишечного тракта. При этом злокачественные новообразования, как правило, развиваются вследствие малигнизации полипов. Наиболее известными заболеваниями являются синдром Гарднера, синдром Пейтца—Егерса, болезнь Кауден. синдром Мюир—Торре, синдром Хоуэлл—Эванса, множественная эндокринная неоплазия III типа.
Синдром Гарднера
Синдром Гарднера — наследственный симптомокомплекс, включающий различные кожные и костные проявления в сочетании с предраковым интестинальным полипозом толстой кишки. Тип наследования — аутосомно-доминантный с различной степенью экспрессивности гена, локализованного в хромосоме 5. Первый постоянный признак синдрома, проявляющийся в возрасте от 4 до 10 лет (редко позднее), — эпидермальные и сально-железистые кисты, десмоидные опухоли, фибромы, липомы, трихоэпителиомы, кератоакантомы, лейомиомы, особенно на коже лица, реже — на волосистой части головы, конечностях, груди. Остеомы развиваются главным образом в челюстных и клиновидных костях (в 50% случаев), размеры их небольшие, опухоли чаще множественные.
Железистые полипы при синдроме Гарднера различных отделов толстой или только прямой кишки развиваются на 3-4-м десятилетии жизни и могут оставаться бессимптомными до тех пор, пока не произойдет их озлокачествление. Гистологически фокусы злокачественной трансформации выявляются в 100% полипов, однако клинически ее можно заподозрить у 50-100% больных. Почти в 50% случаев отмечается полипоз желудка и тонкой кишки, в частности двенадцатиперстной кишки.
Иногда при синдроме Гарднера наблюдаются фибросаркома, лейомиома желудка или кишечника. Также описаны опухоли щитовидной железы, яичников, надпочечников, печени, меланома
Кожные проявления при синдроме Гарднера обычно развиваются задолго до полипоза кишечника, тем самым облегчая его распознавание.
Диагноз синдрома Гарднера основывается на клинических данных и результатах специальных методов исследования пищеварительного тракта — повторных колоноскопий. Диагностическую значимость также имеет врожденная гипертрофическая пигментация сетчатки.
Дифференциальный диагноз синдрома Гарднера проводится с болезнью Кауден, синдромами Пейтца—Егерса, Кронкхайма—Канада, Мюир—Торре.
Лечение синдрома Гарднера заключается в раннем профилактическом удалении полипов толстой кишки.
Синдром Пейтца—Егерса
Синдром Пейтца—Егерса (син.: периорифициальный лентигиноз) — заболевание, проявляющееся пигментными пятнами, сопровождающимися гамартомами желудочно-кишечного, дыхательного и мочеполового трактов. Мужчины и женщины болеют одинаково часто. Тип наследования аутосомно-доминантный. Локус гена неизвестен. Вероятно, заболевание обусловлено мутацией одного плейотропного гена. Может проявляться в детстве, но чаще всего изменения возникают в юности или ранней молодости. Злокачественные опухоли при синдроме Пейтца—Егерса встречаются в раннем возрасте, частота их составляет 44-48%.
Большинство больных (95%) синдромом пейтца-егерса имеют характерные пигментные пятна (лентиго, веснушки) темно-коричневого цвета, круглой или овальной формы, диаметром от 2 до 5 мм на губах (особенно на нижней) или слизистой оболочке щек, а также вокруг рта и на переносице, в области заднего прохода, реже на кистях и стопах, ладонях, подошвах, в подколенных ямках. Также описаны пигментные папилломы слизистой оболочки рта. Иногда наблюдается выпадение волос. Очаги пигментации могут быть врожденными, появляться в младенчестве или детстве и со временем бледнеют, хотя пигментация на слизистой оболочке сохраняется.
Гистологически при синдроме пейтца-егерса в эпидермисе обнаруживают увеличение количества меланоцитов в базальном слое, в дерме — скопление меланофоров.
Гамартомы при синдроме пейтца-егерса развиваются в тонкой кишке, хотя могут встречаться в любом отделе желудочно-кишечного тракта, а также в желчевыводящих путях, дыхательном и мочеполовом трактах. Они имеются у подавляющего большинства больных и представляют собой полиповидные образования небольших размеров округлой формы с гладкой поверхностью, сопровождаются приступами болей в животе и желудочно-кишечными кровотечениями. Гистологически полипы имеют строение доброкачественной аденомы, в 20-25% случаев они подвергаются озлокачествлению. Однако злокачественные опухоли желудочно-кишечного тракта встречаются при этом синдроме только в 2-12% случаев. Значительно чаще наблюдаются новообразования, располагающиеся вне желудочно-кишечного тракта: злокачественные опухоли половых органов (яичников, яичек), рак легких и молочной железы и др.
Диагноз синдрома Пейтца—Егерса устанавливается на основании клинической картины и результатов гистологического исследования. В целях раннего выявления полипоза проводят рентгенологическое и эндоскопическое обследование больных.
Дифференциальный диагноз синдрома Пейтца—Егерса проводится с веснушками, старческим лентиго, синдромом LEOPARD, наследственными формами лентигиноза, особенно системного, а также с мастоцитозом.
Синдром Пейтца—Егерса нередко заканчивается летальным исходом, обусловленным несвоевременно распознанными злокачественными новообразованиями внутренних органов.
Лечение синдрома Пейтца—Егерса пигментации губ проводят лазерным облучением. Полипы, превышающие диаметр 1,5 см, а также кровоточащие полипы удаляют хирургически. Каждые 1-3 года больной должен осматриваться гастроэнтерологом и хирургом. Иногда показана профилактическая колэктомия.