Муковисцидоз: что за болезнь, как проходит лечение?
Муковисцидоз — это наследственное заболевание, которое обычно проявляется в первые 2 года жизни ребенка. Муковисцидоз затрагивает практически все органы, которые выделяют слизь: легкие, печень, поджелудочную железу, кишечник.
Выделяемые секреты становятся густыми, накапливаются в бронхах и железистых протоках, что ведет к формированию кист (новообразований в виде полостей, заполненных жидкостью или воздухом), поэтому болезнь называют иногда кистозным фиброзом 1 .
Трудности диагностики МПС
МПС относятся к очень редким (орфанным) заболеваниям. В разных странах различные МПС регистрировали с частотой 1 на 16000-29000 живых новорожденных [6,7], а в 2007 году в Скандинавских странах распространенность МПС составила всего 4-7 случаев на 1 млн населения [8]. В связи с этим информированность врачей, особенно наблюдающих взрослых пациентов, о МПС низкая. Дополнительные сложности в диагностике возникают при более легком течении МПС, особенно при отсутствии типичных фенотипических проявлений, таких как низкий рост и характерные черты лица. Например, в зависимости от клинических проявлений и течения выделяют три формы МПС I – тяжелую (синдром Гурлера), промежуточную (синдром Гурлера-Шейе) и более легкую (синдром Шейе). Во всех случаях причиной заболевания является мутация гена, кодирующего α-L-идуронидазу. У пациентов с синдромом Гурлера симптомы появляются в раннем детском возрасте и часто наблюдается тяжелое поражение ЦНС, в то время как при синдроме Шейе симптомы менее выражены и возникают значительно позднее, а когнитивные расстройства обычно отсутствуют [9]. Два варианта течения заболевания – тяжелый и более легкий – возможны и при МПС VI (синдроме Марото–Лами), обусловленном мутациями гена, кодирующего арилсульфатазу В.
В клинике им. Е.М. Тареева за последние 3 года были обследованы 5 взрослых пациентов в возрасте от 20 до 33 лет с МПС VI. У трех из них диагноз был установлен в подростковом возрасте (от 7 до 16 лет), а у двух – в возрасте 23 и 30 лет, соответственно. Необ хо димо подчеркнуть, что хотя у двух последних пациенток наблюдалось замедленное прогрессирование заболевания, тем не менее, в обоих случаях на момент госпитализации в клинику имелось тяжелое поражение опорно-двигательного аппарата с резким ограничением подвижности в суставах, пороки клапанов сердца, стеноз шейного отдела позвоночника, нарушение проходимости дыхательных путей, поражение органа зрения и др. Обе пациентки были низкого роста (132 и 146 см) [4].
Замедленное прогрессирование течение иногда на блюдается и при МПС II (синдроме Хантера). Два года назад в нашу клинику был госпитализирован 42-летний пациент с МПС II, который был диагностирован в возрасте 13 лет на основании характерных изменений внешнего вида и наличия синдрома Хантера у старшего брата и подтвержден при энзимологическом (дефицит активности идуронат-2-сульфатазы) и молекулярногенетическом (мутация с.236С>А гена IDS в гемизиготном состоянии) исследованиях [10]. В течение длительного времени состояние пациента оставалось удовлетворительным. Успешно закончил школу, а затем институт. Работал инженером на заводе. С 30-летнего возраста прогрессирующее снижение чувствительности и боли в кистях и стопах, ухудшение зрения и выпадение центральных полей зрения, однако продолжал работать. Резкое ухудшение состояния, связанное с развитием сердечной недостаточности на фоне тяжелого порока аортального клапана, было отмечено только за год до госпитализации, т.е. в возрасте около 40 лет.
При обращении к ревматологу на МПС может указывать поражение суставов, не сопровождающееся признаками воспаления, такими как припухание, повышение СОЭ и/или уровня С-реактивного белка [12]. T. Rocha Siqueira и соавт. измеряли экскрецию ГАГ с мочой у 55 пациентов в возрасте от 3 до 21 года (в среднем 9 лет) с невоспалительной артропатией неясного генеза. У всех больных определялись дискомфорт или боль в суставах, а у 2/3 – скованность [12]. Экскре ция ГАГ была повышена у 1 из 55 больных. При дополнительном обследовании у 15-летней пациентки был установлен диагноз МПС II. Хотя очевидным ограничением этого исследования было небольшое число обследованных пациентов, тем не менее, полученные данные указывают на возможную роль скрининга в диагностике более легких форм МПС.
Симптомы мукополисахаридоза
Мукополисахаридозы делятся на несколько типов. Они различаются первичным генным дефектом, патологическим ферментом, преимущественным поражением той или иной системы органов и тканей, возрастом начала заболевания и тяжестью его течения (см. классификацию ). В целом при этих генетических патологиях встречаются множественные нарушения: поражаются костная система, хрящи, печень, селезёнка, головной мозг, роговица глаза, органы лимфатической и дыхательной системы. Из-за особенностей строения дыхательных путей возникают частые инфекционные заболевания органов дыхания и слуха, что приводит к развитию тугоухости и респираторным расстройствам — бронхитам, пневмониям и др.
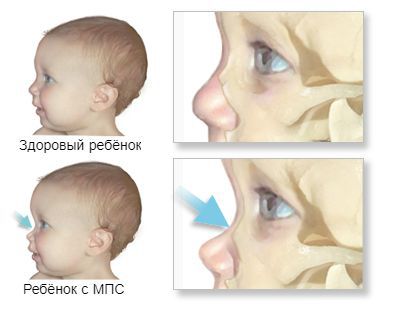
К самым частым проявлениям мукополисахаридозов относят низкорослость, волосатость и нарушения развития. Они дают о себе знать с раннего детства. Лицо ребёнка, как правило, приобретает грубые черты, становится «взрослым», а голова выглядит довольно большой из-за широкого лба и короткого носа. Губы и язык тоже становятся больше, чем у сверстников. Суставы пальцев плохо гнутся и двигаются. При отдельных типах заболевания нарушается слух и зрение, повреждаются сердечные клапаны и артерии [14] .
Шаг 2. Исключение других болезней накопления
Ряд болезней накопления имеет схожие с МПС внешние клинические признаки. Например, симптоматика альфа-маннозидоза включает:
- грубые черты лица;
- изменения скелета по типу множественного дизостоза;
- поясничный лордоз;
- диффузную мышечную гипотонию;
- шумное носовое дыхание;
- умеренную гепатоспленомегалию;
- нейросенсорную тугоухость;
- пупочную, паховую грыжи;
- умственную отсталость различной степени;
- частые респираторные инфекции, бронхиты, отиты.
Но для альфа-маннозидоза ведущим клиническим признаком является высокая степень нейросенсорной тугоухости, наличие признаков периферической полинейропатии (для МПС-1, МПС-2, МПС-6 характерен только синдром запястного канала), гуморальные и клеточные иммунодефициты. У пациентов с МПС, несмотря на то, что они склонны к частым респираторным инфекциям, иммунодефициты отсутствуют. Но главное отличие в результатах лабораторных исследований. При альфа-маннозидозе отмечается снижение активности альфа-маннозидазы в лейкоцитах. Накопление маннозосодержащих олигосахаридов. ГАГ в моче отсутствует. По результатам ДНК-диагностики заболевание обусловлено мутациями в гене MAN2B1.
Муколипидоз 2-го и 3-го типа также имеет симптомы, которые «роднят» его с разными типами МПС. Муколипидоз 2-го типа имеет более тяжелое течение и клинические проявления практически с первых месяцев жизни, быстрое прогрессирование. Муколипидоз 3-го типа мягче, отличается более поздним проявлением симптоматики и чуть меньшей скоростью прогрессирования. При этом вне зависимости от типа муколипидозу свойственны общие с мукополисахаридозом симптомы:
- грубый гаргоилизм;
- низкорослость;
- помутнение роговицы;
- тугоподвижность суставов;
- интеллектуальный дефицит;
- паховые и пупочные грыжи;
- органомегалия и др.
Отличить одно заболевание от другого помогает лабораторная диагностика. При муколипидозе 2-го типа отмечается повышение уровня лизосомных ферментов (N-ацетилгексозаминидазы) в плазме и низкий уровень в лейкоцитах крови. При муколипидозе 3-го типа — резкое снижение активности N-ацетилглюкозамин-1-фосфотрансферазы в лейкоцитах и культуре кожных фибробластов (ККФ). В отличие от МПС муколипидоз обусловлен мутациями в гене GNPTAB.
Фукозидоз — очень редкая патология, которая имеет общие с МПС симптомы: огрубление черт лица, низкорослость, органомегалию, грыжи, нейросенсорную тугоухость, контрактуры крупных и мелких суставов, помутнение роговицы и т. д.
Как и другие болезни накопления, фукозидоз может протекать с разной скоростью прогрессирования. При тяжелой форме болезни клинические проявления возникают уже на первом году жизни, быстро прогрессируют, приводя к тяжелой инвалидизации и раннему летальному исходу. При мягком течении клинические симптомы появляются в конце первого десятилетия, медленнее прогрессируют.
Главной отличительной особенностью фукозидоза являются специфические кожные проявления — ангиокератома, ангидроз. Наличие ангиокератомы вкупе с характерной для МПС симптоматикой дает основания предполагать фукозидоз. Окончательное слово за лабораторной диагностикой. При фукозидозе отмечается резкое снижение активности α-L-фукозидазы и ККФ, накопление гликолипидов и гликопротеинов в клетках. Молекулярно-генетическая диагностика указывает на мутации в гене FUCA1.
GM1-ганглиозидоз , в отличие от предыдущего заболевания, широко распространен среди болезней накопления. Несмотря на некоторые схожие с МПС симптомы (гаргоилизм, низкорослость, гиперплазия десен, костные деформации, органомегалия, тугоподвижность суставов и т. д.), GM1-ганглиозидоз имеет яркие отличительные особенности. В частности, ему свойственны макулодистрофия по типу «вишневой косточки», поражение почек, быстро прогрессирующее поражение ЦНС, эпилепсия, грубая задержка психомоторного развития, ангиокератома. По результатам лабораторной диагностики отмечается резкое снижение активности β-галактозидазы в лейкоцитах и ККФ. Заболевание обусловлено мутациями в гене GLB1. Болезнь практически всегда имеет тяжелое течение и неблагоприятный исход в первые годы жизни.
Множественная сульфатазная недостаточность — заболевание, которое легко спутать с МПС 2-го типа, если не выполнить объективную лабораторную диагностику. Клиническая симптоматика заболевания почти идентична симптоматике МПС-2, но результаты лабораторных исследований показывают принципиальные отличия. Так, при множественной сульфатазной недостаточности в крови снижен уровень не только идуронат-2-сульфатазы, но и других ферментов, в частности арилсульфатаз А, В, С. ДНК-диагностика показывает мутации в гене SUMF1.
Патологические изменения, лежащие в основе заболевания.
Мукополисахаридозы представляют собой группу из 11 редких наследственных (т.е. передающихся от родителей к детям) заболеваний, которые связаны с нарушением обмена гликозаминогликанов. Данные вещества присутствуют в организме человека повсеместно и заполняют пространство между клетками, поэтому нарушение их метаболизма приводит к серьезным нарушениям функций практически каждого органа. В зависимости от гена, который поражается при мукополисахаридозе, определяется тип заболевания. Всего выделяют 11 генов, поломки (мутации) в которых приводят к снижению или потере функции определенных ферментов, разрушающих гликозаминогликаны и приводящих к развитию мукополисахаридоза какого-либо типа. В настоящее время только 4 из 11 типов мукополисахаридозов поддаются лечению: мукополисахаридоз I, II, IV и VI типов. Своевременно начатая терапия позволяет улучшить качество жизни заболевшего и предотвратить серьезные нарушения функций организма.
Мукополисахаридоз I типа (МПС I) – как и другие типы мукополисахаридозов, является наследственным заболеванием, причиной которого является унаследованный от обоих родителей (в большинстве случаев, здоровых) поломанные гены. Из-за генных изменений возникает дефицит фермента (альфа-L-идуронидаза), приводящий к накоплению мукополисахаридов (или гликозаминогликанов) в соединительной ткани всех органов – в центральной нервной системе, сердце, сосудах, опорно-двигательном аппарате, печени, селезенке, органах зрения и слуха.

В зависимости от проявлений заболевания выделяют разные по степени тяжести формы МПС I (см. Признаки и симптомы мукополисахаридоза) 10
Лечение и профилактика родинки-меланомы
В настоящее время в косметологических салонах предлагают бесследное удаление родинок современными методами. Этого делать не стоит. Любую родинку должен исследовать и удалять онколог. Удалению подлежат родинки, которые часто травмируются, расположены на открытых участках тела, где высока опасность солнечного воздействия. Онкологи Юсуповской больницы при удалении родинки и признаками трансформации в меланому строго соблюдают принципы абластики.
Комплекс мер по предупреждению распространения во время операции атипичных клеток состоит из следующих мероприятий:
- Разрезы выполняют только в пределах заведомо здоровых тканей;
- Избегают механического повреждения ткани новообразования;
- Как можно быстрее перевязывают венозные сосуды, которые отходят от новообразования;
- При наличии такой возможности накладывают жгут выше и ниже опухоли для предупреждения миграции раковых клеток;
- Перед манипуляциями с родинкой, перерождённой в меланому, ограничиваю рану салфетками;
- После удаления новообразования меняют инструменты, перчатки и ограничивающие салфетки.
На начальных этапах родинки-меланомы удаление новообразования осуществляется следующими методами:
- Электрокоагуляцией – с помощью тока производится «выпаривание» родинки (процедура достаточно болезненна и имеет высокий риск появления рецидива опухоли);
- Разрушением жидким азотом – под воздействием низкой температуры происходит разрушение клеток родинки, что приводит к тому, что она отпадает самостоятельно. После процедуры не остается рубцов, но она эффективна только при наличии поверхностных родинок;
- Лазерной вапоризацией – под воздействием направленного лазерного луча содержимое родинки испаряется. Процедура эффективна, позволяет удалить сразу несколько новообразований за один раз.
Лазерным лучом не удаляют родинки, размер которых превышает 2 см. В настоящее время не разработаны меры специфической профилактики озлокачествления родинок. Предотвратить развитие меланомы позволяет соблюдение следующих рекомендаций:
- Проведение регулярных самоосмотров;
- Использование солнцезащитных средств;
- Удаление новообразований, которые расположены в зоне постоянного раздражения;
- Прохождение профилактических осмотров у дерматолога.
Перерождение родинки в меланому – процесс, который при внимательном отношении к своему организму сложно пропустить. Если вы заметили изменения в родинке, звоните в контакт центр Юсуповской больницы и записывайтесь на приём к дерматологу. При своевременной диагностике и проведении адекватной терапии на начальных этапах трансформации прогноз для выздоровления улучшается.