Болезнь Реклингхаузена (Нейрофиброматоз)
Институт хирургии им. А.В. Вишневского, Москва
Институт хирургии им. А.В. Вишневского, Москва, Россия
Институт хирургии им. А.В. Вишневского Минздрава РФ, Москва
Институт хирургии им. А.В. Вишневского, Москва
Хирургическое лечение нейрофиброматоза I типа, ассоциированного с опухолевым поражением забрюшинного пространства
Журнал: Хирургия. Журнал им. Н.И. Пирогова. 2019;(3): 5‑14
Берелавичус С.В., Стручков В.Ю., Сон А.И., Кригер А.Г. Хирургическое лечение нейрофиброматоза I типа, ассоциированного с опухолевым поражением забрюшинного пространства. Хирургия. Журнал им. Н.И. Пирогова. 2019;(3):5‑14.
Berelavichus SV, Struchkov VYu, Son AI, Kriger AG. Surgical treatment of neurofibromatosis type I followed by retroperitoneal tumor (in Russian only). Pirogov Russian Journal of Surgery = Khirurgiya. Zurnal im. N.I. Pirogova. 2019;(3):5‑14. (In Russ., In Engl.).
https://doi.org/10.17116/hirurgia20190315
Институт хирургии им. А.В. Вишневского, Москва
Нейрофиброматоз I типа (болезнь Реклингхаузена) — наследственное (аутосомно-доминантное заболевание), вызванное мутацией гена NF1. Опухоли брюшной полости и забрюшинного пространства при болезни Реклингхаузена встречаются редко. Цель исследования — выбор оптимального метода хирургического лечения у пациентов с проявлениями болезни Реклингхаузена в брюшной полости и забрюшинном пространстве. Материал и методы. В НМИЦ хирургии им. А.В. Вишневского хирургическое лечение выполнено 4 пациентам (3 женщины и 1 мужчина) с нейрофиброматозом I типа. Результаты. Выполнены 3 робот-ассистированные операции по удалению 2 неорганных забрюшинных опухолей (плексиформная нейрофиброма и шваннома) и 1 феохромоцитомы правого надпочечника. В исследование также включено одно клиническое наблюдение традиционного хирургического вмешательства по поводу множественных гастроинтестинальных стромальных опухолей двенадцатиперстной и тонкой кишки на фоне нейрофиброматоза I типа. В одном случае послеоперационный период осложнился формированием жидкостного скопления в ложе удаленной опухоли, что потребовало проведения дренирования под УЗ-контролем. В остальных случаях послеоперационный период протекал без осложнений. Заключение. Проведение робот-ассистированных операций в лечении проявлений нейрофиброматоза I типа в забрюшинном пространстве и брюшной полости безопасно и эффективно у пациентов с болезнью Реклингхаузена при наличии единичных образований. При множественных опухолях, расположенных в разных отделах забрюшинного пространства или брюшной полости, целесообразно выбирать традиционный вариант оперативного вмешательства.
Институт хирургии им. А.В. Вишневского, Москва
Институт хирургии им. А.В. Вишневского, Москва, Россия
Институт хирургии им. А.В. Вишневского Минздрава РФ, Москва
Институт хирургии им. А.В. Вишневского, Москва
Нейрофиброматоз I типа (болезнь Реклингхаузена) — наследственное (аутосомно-доминантное заболевание), вызванное мутацией гена NF1. Наиболее частыми клиническими проявлениями являются пигментации кожи цвета кофе с молоком, узелки Лиша (белесоватые пятна — гамартомы) на радужке глазного яблока, а также множественные доброкачественные нейрофибромы кожи и подкожной клетчатки [1—4]. Частота встречаемости заболевания составляет 1 случай на 2500—3000 человек [5, 6].
Возникновение нейрофиброматозных узлов в брюшной полости и забрюшинном пространстве при болезни Реклингхаузена встречается редко [7]. Наиболее частым из них является плексиформная нейрофиброма (ПН) забрюшинного пространства. В литературе [8—11] описаны единичные случаи ассоциации шванном, феохромоцитом и гастроинтестинальной стромальной опухоли (ГИСО) с нейрофиброматозом I типа.
H. Brems и соавт. [8] в систематическом обзоре указывают на 6% частоту встречаемости ГИСО у больных с нейрофиброматозом I типа. В большинстве случаев они представлены множественными опухолями тонкой кишки.
При наличии таких клинических проявлений, как феохромоцитома и ГИСО, пациентам показано удаление новообразований. ПН и шванномы рекомендуется удалять при наличии болевого синдрома, неврологической симптоматики и появлении признаков малигнизации [7].
Патофизилогия и причины
Нейрофибраматоз Реклингаузена появляется в результате мутационных изменений гена NF-1 в семнадцатой q-хромосоме. По данным научных исследований выявлено, что данный ген принимает участие в процессе синтезирования белка нейрофибромина, который отвечает за подавление опухолей.
Если будет недостаточный уровень или полное отсутствие этого гена, то в результате будет наблюдаться переход клеток нервной ткани в состояние опухолей.
Наследование болезни происходит аутосомно-доминантным способом. Если у родителя имеется это нарушение, то вероятность рождения ребенка с данным заболеванием может доходить до 50 %.
Иногда данное заболевание может развиваться в результате спонтанных мутационных изменений, развитие которых наблюдается уже в генетически заложенном материале (яйцеклетке или сперматозоиде) родителей, не имеющим в роду данного отклонения.
Причины нейрофиброматоза Реклингхаузена
Нейрофиброматозы являются наследственными заболеваниями, возникающими в связи с генными нарушениями. Форма Реклингхаузена передается аутосомно-доминантно. Это значит, что вероятность рождения ребенка с патологией у больного родителя составляет 50%. В некоторых случаях нейрофиброматоз Реклингхаузена развивается как результат спонтанной мутации в семье, где случаи этого заболевания ранее не наблюдались.
Клиническая картина нейрофиброматоза Реклингхаузена отличается значительным многообразием симптомов. Самым ранним признаком, позволяющим заподозрить заболевание, является наличие у новорожденного или ребенка более 5-ти пигментных пятен диаметром 1,5 см и больше. Обычно они расположены на туловище и шее, но могут быть на лице и конечностях. Как правило, пигментные пятна при нейрофиброматозе имеют молочно-кофейную окраску, но могут встречаться фиолетовые, синие и даже депигментированные пятна.
Периферическая форма характеризуется развитием множественных неврином периферических нервов и нейрофибром в виде подкожных узелков. Опухоли могут локализоваться на туловище, шее, голове, конечностях. Они имеют округлую форму, подвижны и безболезненны при прощупывании. Размер таких образований в среднем 1-2 см, но в отдельных случаях они достигают значительной величины с весом около 2 кг. Кожа над опухолями часто более пигментированна, на верхушке образования может отмечаться рост волос.
При появлении неврином на терминальных ветвях нервных стволов приводит к затруднению лимфооттока и развитию «слоновости» (лимфедемы), которая проявляется увеличением объема конечности, части лица, языка. Часто НФ1 сочетается с аномалиями развития скелета: сколиозом, асимметрией черепа, незаращением дужек позвонков и др. Специфическим признаком нейрофиброматоза Реклингхаузена 1 типа является обнаружение на радужке пациента беловатого пятнышка — узелка Лиша. Этот признак встречается у 94% пациентов с НФ1.
Для центральной формы нейрофиброматоза Реклингхаузена характерными являются менингиомы или глиомы, поражающие спинномозговые корешки и черепно-мозговые нервы, реже локализующиеся внутри позвоночного канала или черепа. Симптомы поражения ЦНС при этом зависят от расположения опухоли и степени сдавления спинного или головного мозга. Это могут быть головные боли, потеря чувствительности, нарушения равновесия, признаки поражения черепно-лицевых нервов, нарушения речи. При этом, как правило, нарушения двигательной функции отсутствуют.
Примерно у пятой часто больных отмечается поражение зрительного анализатора, проявляющееся развитием нейрофибром оболочек глаза, глаукомы, экзофтальма, опухолью зрительного нерва. Чаще всего при НФ2 наблюдается двусторонняя потеря слуха, обусловленная развитием неврином обоих слуховых нервов. В некоторых случаях это является единственным проявлением заболевания. Нейрофиброматоз Реклингхаузена может сопровождаться умственной отсталостью, эпилептическими приступами, гинекомастией, преждевременным половым созреванием.
Причины
Этиологическим фактором нейрофиброматоза 1 типа являются множественные мутации одного из самых протяженных и сложно организованных генов NF1 (17q11.2), являющихся следствием инактивации преимущественно отцовской аллели. Причиной нейрофиброматоза 2 типа является генетический дефект в 22 хромосоме (22q12).
Нейрофиброматоз Реклингхаузена начинает проявляться в большинстве случаев множественными нейрофибромами, с локализацией по ходу периферических нервов, в виде округлых болезненных узелков, расположенных в толще кожи, различающиеся по своим размерам. Нейрофибромы представлены в виде узелков на/в толще кожи нормальной или синюшно-красной окраски и мягко эластической консистенции.
Характерным признаком, начиная с периода новорожденности/первых лет жизни детей является появление на коже мелких «кофейных пятен», напоминающих веснушки с четкими границами и ровными краями с преимущественной их локализацией на участках тела со складками (в паховой области, подмышечных впадинах, на животе). По мере взросления ребенка наблюдается тенденция к увеличению их численности (от единичных пятен до нескольких сотен) и размеров (от точечных до 2-5 сантиметров в диаметре), они также могут приобретать темную окраску. Следует учитывать, что к диагностически значимым пятнам относятся у детей до пубертата — диаметром более 5 мм и после полового созревания — 15 мм и более. Нейрофиброматоз (фото начальной стадии) приведены ниже.
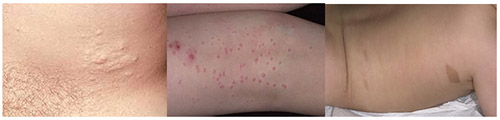
Мягкие кожные опухоли, как правило, у детей отсутствуют, особенно первых лет жизни и возникают к 10-15 годам. При этом, их количество особенно возрастает в пубертатном периоде. При пальпации они безболезненны, однако при вовлечении в патологический процесс периферических нервов, могут возникают боли, гипестезии. На коже могут присутствовать и другие изменения: участки депигментации, сосудистые пятна, очаговое поседение волос, гипертрихоз. Возможно развитие нейрофибром. Плексиформная нейрофиброма может образовывать гигантские опухоли, состоящие из кожных и подкожных элементов.
Некоторые авторы (В.В. Мордовцева) выделяет четыре варианта клинического течения нейрофиброматоза I типа:
- с наличием незначительных пигментных пятен типа веснушек в сочетании с крупными «кофейными» пятнами при отсутствии нейрофибром или с наличием единичных опухолей;
- с наличием преимущественно крупных пигментных кофейных пятен на коже и небольшим количеством нейрофибром;
- с преобладанием нейрофибром и незначительным количеством пигментных пятен (по типу веснушек/крупных);
- смешанный тип.
Факультативные симптомы нейрофиброматоза проявляются изменениями в костной системе в виде различных деформаций позвоночного столба — сколиозы, лордосколиозы, кифосколиозы, псевдоартрозы, утончение/искривление длинных трубчатых костей, патологических переломов. Нередко развиваются лицевые дисморфии: аномалии глазных щелей, глазной гипертелоризм, деформация ушных раковин, неправильная форма черепа и др. Для пациентов с НФ-I характерны макроцефалия, низкий рост, дисплазия клиновидной кости и затылочной части черепа.
Характерными поражениями нервной системы являются судорожный и гипертензионно-гидроцефальный синдромы, опухоли ЦНС (эпендимома, астроцитома, глиома зрительного нерва/ствола мозга), эпилепсия, что появляется соответствующей симптоматикой. Так, глиома зрительного нерва проявляется нарушением зрения.
Для NF1 характерны дополнительные проявления в виде когнитивных расстройств от легких до выраженных, часто в сочетании с умеренным снижением IQ ребенка, затруднением в освоении чтения, письма, математики и нервно-психической неуравновешенностью; наличие эндокринных расстройств (полового созревания, феохромоцитома, нарушение роста и др.). Со стороны сосудов у пациентов часто регистрируется стеноз почечных артерий с повышением АД, окклюзия артерий, болезнь Мойа-Мойа.
Клиническая манифестация заболевания НФ2 включает развитие двусторонних вестибулярных шванном или шванном других черепных периферических и спинальных нервов при минимальных экстраневральных и кожных симптомах. Двусторонняя шваннома (невринома) слухового нерва относится к основным признакам, который встречается практически в 90% случаев, редко обнаруживается у детей, развиваются преимущественно в позднем подростковом/раннем взрослом возрасте. НФ-II.
Значительно реже (около 10%) случаев встречаются различного рода менингиомы (спинальные, интракраниальные, менингиомы зрительных нервов), глиомы и эпиндимомы. Первым симптомом часто является снижение/потеря слуха, появление в ушах шума, который может сопровождаться атаксией и головокружением. Пятна на коже цвета «кофе с молоком» в большинстве случаев отсутствуют или присутствуют в небольшом количестве. У 50-70% больных нейрофиброматозом 2 типа диагностируются зрительные нарушения (гемартромы, катаракты, ретинобластомы, менингиомы зрительных нервов).
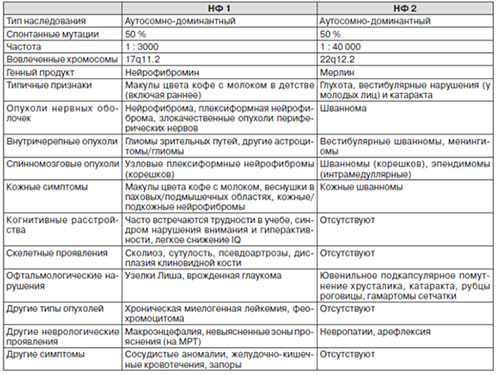
Ниже в таблице приведены характерные проявления нейрофиброматоза 1 и 2 типа.
Последние новости
Совсем недавно стартовала уникальная программа Союзного государства помощи детям с патологиями и деформацией позвоночника, разработанная ведущими специалистами в области вертебрологии из России и Беларуси.
Сегодня эти планы воплотились на практике, подарив первым маленьким пациентам здоровое будущее, без болей и ограничений.
В выпуске новостей телеканала Диёр 24 вышел репортаж из Наманганской области республики Узбекистан о визите в областную больницу врачей-травматологов из НИИ Травматологии и ортопедии, г. Ташкент, и профессора С.В. Виссарионова из НИИ детской ортопедии, г. Санкт-Петербург.
28 февраля 2018 Подробнее
г. Пушкин (пригород г. Санкт- Петербурга), Парковая улица 64-68, ФГУ «НИДОИ им. Г.И.Турнера»
Эндокринные нарушения
Эндокринные нарушения проявляются акромегалией, сахарным диабетом, гипогонадизмом или, наоборот, преждевременным половым созреванием. Наблюдают недостаточность надпочечников, щитовидной железы, кретинизм, гиперпаратироидизм, но самой частой патологией считают феохромоцитому.
Возможны поражения сердечно-сосудистой системы (врожденные пороки сердца, гипертоническая болезнь; коарктация аорты, стеноз легочной артерии) и мочеполового тракта (фиброматоз мочевого пузыря, стеноз почечных артерий). У 10–20% взрослых диагностируют болезни легких, примерно у 10% — невриномы желудка и кишечника. К характерным проявлениям болезни относят диффузную хроническую гепатопатию.
Диагностическим критерием заболевания типа I считают наличие двух или более признаков из указанных ниже (ВОЗ, 1992 г.):
- шесть или более пятен цвета «кофе с молоком» диаметром более 5 мм в препубертатном периоде или диаметром 15 мм в постпубертатном периоде;
- две или более нейрофибромы любого типа либо одна плексиформная нейрофиброма;
- мелкие пигментные пятна в подмышечных и паховых областях, напоминающие веснушки;
- глиома зрительного нерва;
- два или более узелка Лиша (гамартом радужной оболочки);
- костные изменения: дисплазия крыла клиновидной кости, истончение кортикального слоя трубчатых костей с псевдоартрозом или без него;
- наличие нейрофиброматоза типа I у родственников первой степени родства (по этим критериям).
При гистологическом исследовании в пигментных пятнах определяются гигантские гранулы пигмента (макромеланосомы) и DOPA-положительные меланоциты.Нейрофибромы — неинкапсулированные опухоли, в них определяется пролиферация веретенообразных клеток с ядрами волнистых очертаний, пролиферация фибробластических элементов, большое количество незрелых коллагеновых волокон, тонкостенные сосуды, остатки нервных пучков, тканевые базофилы. В стадии активного роста нейрофибром накапливаются кислые мукополисахариды, выявляется большое число тучных клеток в стадии дегрануляции.
При нейрофиброматозе типа I возможна пренатальная диагностика, а также предимплантационная диагностика в процессе проведения ЭКО для семейных пар с высоким риском передачи патологического гена.
- Нейрофибромы могут быть составной частью так называемого NAME-синдрома (син.: синдром Карни), проявляющегося:
- множественными пигментными невусами;
- пигментными пятнами типа веснушек;
- миксомами предсердия;
- миксоидными подкожными нейрофибромами.
- низкий рост;
- короткая шея;
- аномалии грудной клетки и позвоночника;
- крыловидная складка на шее;
- дефекты лица (гипертелоризм, птоз, антимонголоидный разрез глаз, эпикантус, косоглазие, миопия, микрогнатия, нарушение прикуса, аномалии ушей).
- возможны крипторхизм, нарушения менструального цикла, врожденные пороки сердца, в основном правых отделов. Характерен гирсутизм, у отдельных больных — низкий рост волос на затылке, деформация локтевых суставов, иногда кистей и стоп; у значительной части больных — умственная отсталость. Больные склонны к келоидным реакциям, периферическим лимфатическим отекам, повышенной растяжимости кожи.
- наличие крупных сегментированных пятен цвета кофе с молоком
- патология костей (например, полиостотическая фиброзная дисплазия)
- эндокринные нарушения
- Треугольное лицо
- Низкий рост
- Асимметрия скелета
- Нарушение полового созревания
- низкий рост
- эритема и телеангиэктазии на лице
- узкое лицо, выступающие нос и уши
- задержка умственного развития
- стеноз легочной артерии
- пигментные пятна, похожие на веснушки, в подмышечных впадинах
- Наличие многочисленных пятен цвета кофе с молоком без других признаков нейрофиброматоза I типа
Заболевание встречается среди представителей всех рас с частотой 1 на 35 000 населения. Наследование аутосомно-доминантное, в половине случаев заболевание развивается в результате спонтанных мутаций.Локус гена — 22q11.21–q13.1.Развитие заболевания связано с инактивацией первичного продукта гена — шванномина (мерлина), предположительно тормозящего опухолевый рост на уровне клеточных мембран.
По клиническим проявлениям центральный нейрофиброматоз близок к классическому варианту, но отличается частотой и выраженностью симптомов, а также сроками их появления. Ведущий признак — двусторонняя невринома слухового нерва, развивающаяся практически у всех носителей гена и в большинстве случаев способствующая потере слуха в возрасте 20–30 лет. Из других симптомов наблюдают:
- эпилептиформные припадки;
- судороги;
- параличи;
- умственную отсталость;
- менингеальные симптомы, обусловленные локализацией неврином в головном и спинном мозге.
Кожные проявления могут быть минимальными. Пигментные пятна встречаются примерно у 42% больных, а нейрофибромы — у 19%. Более характерны болезненные, плотные и подвижные подкожные опухоли — невриномы (шванномы). Описано развитие множественных плексиформных шванном.Узелки Лиша отсутствуют. Возможно раннее развитие катаракты. Прогноз для выздоровления неблагоприятный, его тяжесть определяется степенью вовлечения в процесс внутренних органов и систем.
Диагноз «нейрофиброматоз центрального типа» может быть поставлен при наличии одного из следующих критериев (ВОЗ, 1992):
- Рентгенологически подтвержденная двусторонняя невринома слухового нерва;
- Двусторонняя невринома слухового нерва у родственника первой степени родства и наличие у пробанда какого-либо из признаков:
- односторонняя невринома слухового нерва;
- плексиформная нейрофиброма или сочетание двух иных опухолей: менигиома, глиома, нейрофиброма — независимо от их расположения;
- любая внутричерепная или спинномозговая опухоль.
При гистологическом исследовании шванномы представлены инкапсулированными опухолями, состоящими из вытянутых веретенообразных клеток и фибриллярного эозинофильного межклеточного матрикса. Участки скопления параллельных рядов клеток в виде частокола, отделенные друг от друга бесклеточным пространством, носят название зоны Антони А, при этом формируются характерные тельца Верокая. Участки отечной муцинозной стромы названы зоной Антони В.
Дифференциальную диагностику проводят с другими формами нейрофиброматоза. Возможна пренатальная диагностика нейрофиброматоза типа II, а также предимплантационная диагностика в процессе проведения ЭКО для семейных пар с высоким риском передачи патологического гена.
Основные симптомы нейрофиброматоза типа III, или смешанного нейрофиброматоза, — опухоли ЦНС, развивающиеся у большинства больных в 20–30 лет. Пигментные пятна имеют большие размеры и меньшую интенсивность окраски, нейрофибромы менее многочисленны, узелки Лиша отсутствуют.
Нейрофиброматоз типа IV (вариантный) также сходен с центральным нейрофиброматозом, однако отличается более многочисленными кожными нейрофибромами, высоким риском развития глиомы зрительного нерва, нейролеммом и менингиом.
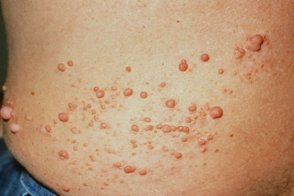
Особо выделяют сегментарный нейрофиброматоз (тип V). Для него характерно одностороннее поражение (нейрофибромы и/или пигментные пятна) какого-либо сегмента тела или его части. Полагают, что развитие этой формы нейрофиброматоза связано с соматической мутацией. Клиническая картина может напоминать таковую при гемигипертрофии; возможно развитие изолированного псевдоартроза конечности; изолированной глиомы зрительного нерва; изолированной плексиформной нейрофибромы. При этом узелки Лиша обнаруживают ипсилатерально. Если они двусторонние, полагают, что данное состояние не отвечает критериям сегментарного нейрофиброматоза. Описан двусторонний сегментарный нейрофиброматоз.
При нейрофиброматозе типа VI нейрофибромы отсутствуют, имеются только пигментные пятна. Диагностика этого заболевания сложна, если не появляются новые случаи в последующих поколениях.
Поздний нейрофиброматоз (тип VII) возникает после 20 лет. По мнению некоторых авторов если процесс развивается в третьей декаде жизни на одном участке тела или более, то он не ассоциирован с пигментными пятнами, узелками Лиша, псевдоартрозом и нарушением умственного развития. Семейные случаи неизвестны.
Отдельные, наиболее крупные нейрофибромы могут быть удалены хирургически. Это показано главным образом при подозрении на злокачественный процесс. Однако после удаления нейрофибром, особенно плексиформных, нередки рецидивы.
Методика лечения включает препараты, влияющие на:
- дегрануляцию тканевых базофилов (кетотифен по 0, 001–0, 002 г 2 раза в день; при выраженном зуде дополнительно назначают фенкарол);
- пролиферацию клеточных элементов (ретиноиды по 1–1, 5 мг/кг);
- снижение количества гликозаминогликанов во внеклеточном матриксе (гиалуронидаза по 64 УЕ через день, на курс 20–30 инъекций).
Продолжительность каждого курса лечения составляет около 3 мес.
Показано медико-генетическое консультирование. Риск унаследовать заболевание для детей больных составляет 50%, независимо от степени клинических проявлений у пробанда. При классическом варианте около 10% носителей гена могут не иметь клинических проявлений. При II типе заболевания пенетрантность более высокая.
Прогноз для выздоровления неблагоприятный, его тяжесть определяется степенью вовлечения в процесс внутренних органов и систем.