Болезнь Марфана: причины, симптомы и лечение

Болезнь (синдром) Марфана – это наследственная патология соединительной ткани. Поскольку соединительная ткань является основой различных органов человека, то и проявления заболевания весьма разнообразны. Самыми типичными являются аномалии сердечно-сосудистой системы, опорно-двигательного аппарата и глаз. Диагностика синдрома Марфана требует использования самых разных методов. Лечение включает в себя медикаментозные и оперативные способы, причем наблюдаться больной должен у нескольких врачей. Из этой статьи Вы сможете узнать о причинах, симптомах, диагностике и вариантах лечения данного заболевания.
Синдром Марфана формируется еще во внутриутробном периоде, ребенок рождается уже с имеющимися нарушениями. Это, пожалуй, наиболее часто встречающаяся наследственная патология соединительной ткани. Независимо от пола и расовой принадлежности, частота выявления болезни Марфана колеблется от 1: 5 000 до 1:10 000 населения (по разным данным).
Причины
Основой заболевания служит генетический дефект: мутация гена, ответственного за синтез белка соединительной ткани фибриллина. Ген расположен на 15-й хромосоме.
Наследуется заболевание по аутосомно-доминантному типу. Аутосомное означает, что оно не связано с полом, а доминантное – то, что мутация всегда проявляется. Степень проявления (распространенность нарушений и их выраженность) может варьироваться в различных пределах, что связано с генетическими особенностями.
Фибриллин придает соединительной ткани эластичность и растяжимость. Нарушение его строения приводит к утрате прочности и упругости соединительной ткани, и она перестает служить прочным каркасом. В первую очередь изменения касаются стенок сосудов, связочного аппарата. Даже небольшие физиологические нагрузки становятся запредельными для организма, соединительная ткань их не выдерживает.
Все случаи синдрома Марфана с точки зрения причины подразделяют на:
- семейные: составляют 75%, это передающаяся из поколения в поколение мутация гена;
- случайные (спорадические): 25%, это впервые возникшая мутация гена в роду, где ранее не было подобной патологии.
Симптомы
Клинических проявлений заболевания весьма много. Их рассматривают с точки зрения поражения отдельных органов и систем:
- опорно-двигательного аппарата;
- сердечно-сосудистой системы;
- органа зрения;
- нервной системы;
- бронхолегочной системы;
- кожи, мягких тканей;
- других органов.
Опорно-двигательный аппарат
Больные с синдромом Марфана обычно имеют высокий рост, худощавы из-за недоразвития подкожно-жировой клетчатки. Туловище при этом кажется коротким, а конечности – непропорционально длинными. Размах рук больше роста на 5% и более. Череп вытянутый (долихоцефалический), пальцы длинные паукообразные (арахнодактилия). Лицо вытянутое, узкое, небо высокое аркообразное (готическое), уши большие, нижняя челюсть выступает вперед (неправильный прикус), зубы растут неправильно, глаза глубоко посажены.
Деформация скелета заключается в наличии воронкообразной или килевидной грудной клетки, любых изменений в оси позвоночника (избыточный лордоз, кифозы, кифосколиозы, сколиозы), подвывихов позвонков (особенно в шейном отделе), спондилолистеза (смещение вышележащего позвонка по отношению к нижележащему). Характерно плоскостопие различных степеней выраженности, разболтанность суставов, избыточные движения в них (например, переразгибание), протрузия вертлужной впадины (углубление с глубоким погружением головки бедренной кости в области тазобедренного сустава). Обычно все это сопровождается снижением мышечного тонуса.
Сердечно-сосудистая система
Изменения в этой системе часто являются наиболее тяжелыми, требующими коррекции в первую очередь, поскольку могут иметь угрожающий жизни характер.
Чаще всего синдром Марфана проявляется наличием пороков развития сердца и крупных сосудов (в частности, аорты). Дефекты строения сердца заключаются в нарушении перегородок сердца и его клапанов (пролапс митрального клапана, дефект межжелудочковой или межпредсердной перегородки, патологическое удлинение хорд). Нарушение строения стенки аорты приводит к развитию ее дилятации (расширению) и расслоению стенок (расслаивающая аневризма аорты). Из врожденных пороков крупных сосудов могут наблюдаться коарктация аорты, стеноз (сужение) легочной артерии.
Патологическое строение сердца и сосудов сопровождается развитием сердечной недостаточности, нарушением сердечного ритма (наджелудочковая и желудочковая тахикардии, фибрилляция предсердий и других), что весьма опасно для жизни больного. Присоединение инфекционных осложнений чревато развитием инфекционного эндокардита.
Орган зрения

Симптомы поражения глаз очень специфичны для данного заболевания. Поражение опорно-двигательного аппарата, сердечно-сосудистой системы и глаз составляют типичную триаду симптомов при болезни Марфана.
Больные синдромом Марфана часто имеют голубые склеры. Зрение плохое ввиду развития близорукости высокой степени, длина глазного яблока увеличена. Зрачки могут быть не симметричными, то есть разными по размеру. Однако этим поражение органа зрения не ограничивается. Развиваются косоглазие, вывих или подвывих хрусталика, недоразвитие радужки и ресничных мышц, уплощение роговицы. Возможна отслойка сетчатки с развитием слепоты.
Нервная система
Поражение нервной системы в основном связано с патологией строения стенки сосудов. Это приводит к нарушению кровообращения, развитию ишемических или геморрагических инсультов (чаще – субарахноидальных кровоизлияний).
Возможны частые обмороки. Из аномалий возможны эктазия твердой мозговой оболочки, в частности пояснично-крестцовое менингоцеле (выпячивание оболочек мозга под кожу с образованием кармана через дефект в позвонках).
Возможны отклонения в психомоторном развитии, причем как в сторону задержки, так и в сторону превышения показателей. Многие люди с синдромом Марфана имеют высокие показатели интеллекта (выше, чем среднестатистический показатель IQ в популяции).
Еще одним симптомом при болезни Марфана могут служить гипофизарные расстройства с повышением содержания адреналина в крови, развитием акромегалии и несахарного диабета.
Бронхолегочная система
Поражение бронхов и легких заключается в развитии спонтанных пневмотораксов, дыхательной недостаточности, эмфиземы легких, апикальных булл (пузырьковидные образования в верхушках легких). Все эти изменения становятся результатом нарушения ангиоархитектоники легких из-за патологии соединительной ткани, повышенной растяжимости и пониженной эластичности легочной ткани.
Поражение кожи и мягких тканей
Часто больные с синдромом Марфана имеют атрофические стрии на коже: волнистые полосы на коже разной ширины и цвета (от белого до красно-фиолетового). Малое количество подкожно жировой клетчатки становится причиной малого веса таких больных.
Больные с синдромом Марфана страдают рецидивирующими паховыми и бедренными грыжами.
Другие органы
Патология соединительной ткани становится причиной опущения почек, мочевого пузыря и матки, варикозного расширения вен, приводит к образованию длинного и со слабой перистальтикой кишечника (что сопровождается запорами).
Диагностика
Диагностика синдрома Марфана требует учета анамнестических данных (в том числе и наследственного анамнеза), данных осмотра больного, а также проведения разнообразных дополнительных методов исследования, позволяющих обнаружить изменения в сердце, сосудах, головном мозге, легких, глазах. Для этого могут понадобиться ЭКГ, ЭхоКГ (УЗИ сердца), рентгенография грудной клетки и тазобедренных суставов, КТ или МРТ головного и спинного мозга, сердца и сосудов, офтальмоскопия, аортография и другие методы. Перечень исследований зависит от наличия симптомов у конкретного больного.
Существуют диагностические критерии этого заболевания: большие и малые. Определенное сочетание больших и малых критериев и позволяет подтвердить диагноз. К большим критериям, например, относят подвывих хрусталика, эктазию твердой мозговой оболочки, килевидную или воронкообразную грудную клетку, плоскостопие, протрузию вертлужной впадины и другие. К малым критериям относят избыточную подвижность в суставах, готическое небо, деформацию черепа, спонтанный пневмоторакс и другие.
Окончательный диагноз выставляется после применения молекулярно-генетического метода и установления мутации гена, ответственного за синтез фибриллина.
Лечение
Лечение синдрома Марфана всегда требует одновременного участия нескольких специалистов: кардиолога, кардиохирурга, офтальмолога, ортопеда-травматолога, терапевта.
Спектр лечебных процедур охватывает консервативные и оперативные способы лечения. Консервативные меры направлены на профилактику осложнений и поддержание нормального функционирования органов и систем, а оперативные предполагают коррекцию имеющихся анатомических изменений с целью предотвратить выраженное нарушение функций или даже угрозу для жизни больного.
Хирургические методы лечения:
- реконструктивные операции на аорте (при значительном, более 5 см, расширении восходящей части аорты и расслоении ее стенки);
- протезирование клапанов сердца;
- удаление измененного хрусталика с заменой его на искусственный;
- пластика позвоночника при выраженном сколиозе;
- эндопротезирование тазобедренных суставов;
- пластика грудной клетки (в последние годы отрицается ее целесообразность).
Больному необходим подбор очков или контактных линз, иногда возможна лазерная коррекция зрения.
Медикаментозное лечение имеет патогенетическую и симптоматическую направленность. Больному с метаболической целью назначают большие дозы витамина С (1-3 г в сутки), препараты с глюкозаминосульфатами и хондроитинсульфатами (Терафлекс, Структум, Хондроксид, Глюкозамин, Аминоартрин, Эльбона, Юниум), Карнитина хлорид 20% раствор, Янтарную кислоту по 100-200 мг 2 раза в день, препараты магния (например, Магне В6), поливитаминно-минеральные комплексы (с кальцием, магнием, цинком, медью). Прием этих веществ направлен на нормализацию обмена веществ, укрепление соединительной ткани.
 Для лечения сердечно-сосудистых нарушений часто используют β-адреноблокаторы (Пропранолол, Обзидан, Атенолол), блокаторы кальция (Нифедипин, Амлодипин, Лекоптин), ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Периндоприл, Эналаприл), антиаритмические препараты (при нарушении ритма сердца). При развитии инфекционного эндокардита показаны антибиотики. После оперативных вмешательств может понадобиться антикоагулянтная терапия для снижения свертываемости крови (Фраксипарин, Клексан).
Для лечения сердечно-сосудистых нарушений часто используют β-адреноблокаторы (Пропранолол, Обзидан, Атенолол), блокаторы кальция (Нифедипин, Амлодипин, Лекоптин), ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Периндоприл, Эналаприл), антиаритмические препараты (при нарушении ритма сердца). При развитии инфекционного эндокардита показаны антибиотики. После оперативных вмешательств может понадобиться антикоагулянтная терапия для снижения свертываемости крови (Фраксипарин, Клексан).
В целом подбор метода лечения и ассортимент применяемых лекарственных средств очень индивидуальны. Все зависит от спектра клинических симптомов и выраженности нарушений у конкретного больного.
Больным с синдромом Марфана показана лечебная физкультура, но в строго дозированном количестве, чтобы занятия приносили пользу сердечно-сосудистой и опорно-двигательной системам, а не вред. Нельзя заниматься контактными и игровыми видами спорта (баскетбол, футбол), рекомендовано плавание.
Прогноз
Заболевание неизлечимо, но постоянное динамическое наблюдение за больным, хирургическая коррекция сердечно-сосудистых, суставных и офтальмологических нарушений позволяют снизить риск для жизни, улучшить ее качество, позволить заниматься своей профессией. Некоторые больные не доживают до 40 лет, что связано с сердечно-сосудистыми осложнениями, в других случаях продолжительность жизни составляет 70 лет (особенно после проведения кардиохирургических операций).
Таким образом, синдром или болезнь Марфана – это наследственное заболевание, течение которого во многом зависит от тщательности медицинского наблюдения и своевременности оказания медицинской помощи. Обычно лечение требует привлечения многих специалистов, но такой комплексный подход позволяет избежать множества осложнений и повысить качество жизни больного.
Синдром Марфана
Синдром Марфана (или арахнодактилия) – это наследственное заболевание, характеризующееся недостаточностью соединительной ткани. Болезнь приводит к патологическим изменениям сердечно-сосудистой, нервной, опорно-двигательной и других систем и органов.
Причины возникновения синдрома Марфана
 Арахнодактилия наследуется по аутосомно-доминантному типу, поэтому встречается почти в одинаковом соотношении, как у мужчин, так и у женщин. Это достаточно редкое генетическое заболевание с частотой возникновения 1:5000.
Арахнодактилия наследуется по аутосомно-доминантному типу, поэтому встречается почти в одинаковом соотношении, как у мужчин, так и у женщин. Это достаточно редкое генетическое заболевание с частотой возникновения 1:5000.
Многочисленные исследования показали, что синдром Марфана обусловлен мутацией в 15-й хромосоме гена белка фибриллина, вследствие чего нарушается структура и выработка коллагена.
По статистике в 75% случаев мутировавший ген передается детям от заболевших родителей, а в остальных случаях к болезни приводят спонтанно возникшие в момент зачатия генетические мутации.
Причины подобных «поломок» в генах до конца так и не выяснены, но с вероятностью в 50% можно утверждать, что дети, рожденные с синдромом Марфана, передадут это заболеванием потомкам.
Признаки синдрома Марфана
К самым заметным симптомам болезни относятся внешние изменения облика человека. Так, заболевшие люди имеют высокий рост относительно своих родственников. Другой признак, бросающийся в глаза – это астеническое телосложение, при котором конечности гораздо длиннее, а также наблюдается арахнодактилия, или «паучьи пальцы». У всех людей с этим заболеванием удлиненный череп, маленькая челюсть с неправильно растущими зубами и глубоко посаженные глаза.
Обычно признаки синдрома Марфана разделяют на группы в соответствии с органами и системами, в которых они проявляются. Например, высокий рост и удлиненные конечности – это проявления со стороны опорно-двигательного аппарата. Также к ним относятся искривление позвоночника, деформация грудной клетки, мягкость суставов и плоскостопие.
Симптомы со стороны глаз – помутнение и вывих хрусталика (его смещение с естественного местоположения), близорукость, отслаивание сетчатки и повышенное внутриглазное давление.
Наиболее опасными признаками синдрома Марфана являются симптомы, касающиеся сердечно-сосудистой системы. Заболевание приводит к расслоению стенок аорты и расширению ее корня, а это чревато неожиданным разрывом главной артерии сердца и практически неминуемым летальным исходом. Кроме того, при болезни Марфана иногда наблюдается неплотное закрытие клапанов сердца, что приводит к увеличению его размеров, шумам, одышке и нерегулярному сердцебиению.
Менее серьезными считаются проявления патологии со стороны легких, кожи и нервной системы.
Диагностика синдрома Марфана
Диагностировать синдром Марфана можно лишь после комплексного обследования специалистами разного профиля: кардиологом, ортопедом, офтальмологом и генетиком.
Врач, специализирующийся на наследственных нарушениях, изучит историю семьи с целью выявления тех родственников, которые умерли от сердечно-сосудистых заболеваний.
Кардиолог назначит рентген грудной клетки и измерение электрической активности сердца (ЭКГ). Также необходима эхокардиограмма, на основе которой будут получены размеры аорты и проверено функционирование клапанов.
Врач-ортопед определит, есть ли искривление грудной клетки и позвоночника, плоскостопие и другие общие проблемы.
Осмотр глаз офтальмологом позволит обнаружить патологические изменения хрусталика, если таковые имеются.
Критерии диагностики синдрома Марфана достаточно строгие и точные. Это связано с тем, что ряд особенностей болезни типичен (например, не всегда высокий и худой человек с длинными тонкими пальцами болен). Кроме того, существуют и другие заболевания соединительной ткани, которые сопровождаются похожими симптомами.
Окончательный диагноз зависит от подтверждения или опровержения арахнодактилии в семейном анамнезе. Если у кого-то из родственников была эта болезнь, достаточно подтверждения в двух системах организма, в противном случае – в трех.
Симптомы заболевания не проявляются в раннем детстве, а обнаруживаются лишь с возрастом. Своевременная диагностика синдрома Марфана очень важна, но даже если диагноз ставится годами, лечить признаки можно еще до окончательного вердикта врачей.
Лечение синдрома Марфана
На сегодняшний день не существует эффективных методик лечения синдрома Марфана. Все мероприятия сводятся к предупреждению осложнений болезни.
 Чтобы предотвратить опасные аортальные изменения, назначается препарат Анаприлин, но его эффективность не доказана. Иногда проводится хирургическая пластика аорты и клапанов сердца. Беременным женщинам с болезнью Марфана и выраженной патологией со стороны сердечно-сосудистой системы проводят плановое кесарево сечение. Для профилактики тромбозов и инфекционного эндокардита после оперативного вмешательства назначаются антикоагулянты и антибиотики.
Чтобы предотвратить опасные аортальные изменения, назначается препарат Анаприлин, но его эффективность не доказана. Иногда проводится хирургическая пластика аорты и клапанов сердца. Беременным женщинам с болезнью Марфана и выраженной патологией со стороны сердечно-сосудистой системы проводят плановое кесарево сечение. Для профилактики тромбозов и инфекционного эндокардита после оперативного вмешательства назначаются антикоагулянты и антибиотики.
При сильном сколиозе показаны физиотерапевтические процедуры, а также механическое укрепление скелета. Хирургическая коррекция проводится в том случае, если угол отклонения позвоночника составляет 45° и более.
Прогноз жизни людей с синдромом Марфана зависит, в первую очередь, от тяжести и степени изменений, затрагивающих сердечно-сосудистую систему. При разрыве легочного ствола и аневризме аорты в большинстве случаев пациент умирает.
Лечение синдрома Марфана подразумевает постоянный врачебный контроль и регулярное диагностическое обследование. Физическая активность больных должна быть снижена до среднего или низкого уровня, то есть следует исключить спортивные соревнования, контактные виды спорта, подводное плавание и изометрические нагрузки. Женщинам детородного возраста, имеющим данное заболевание, необходимо обязательно пройти медико-генетическое консультирование.
Видео с YouTube по теме статьи:
Как лечить паучьи пальцы
В числе серьезных генетических заболеваний находится синдром Марфана, которым страдает один человек из 10-20 тысяч. Им болели такие всемирно известные личности, как Авраам Линкольн, Никколо Паганини, Корней Чуковский и Ганс Христиан Андерсен.
У больного этим синдромом присутствуют различные симптомы: арахнодактилия (удлиненные кисти, паукообразные пальцы рук и ног), множественные костно-суставные аномалии, деформация грудной клетки, нарушения сердечно-сосудистой системы и т.д. В этой статье речь пойдет об одном из них — арахнодактилии, ее проявлениях, методах диагностики и способах лечения.
Что такое арахнодактилия
Это вид наследственной патологии человека, который проявляется аномально длинными и тонкими пальцами рук, их чрезмерной подвижностью, вплоть до сгибания на 180 градусов к тыльной стороне ладони. В народе это заболевание приобрело название болезни «,паучьи пальцы»,.
После описанного впервые синдрома Марфана в 1876 году было выявлено, что «,паучьи пальцы», могут быть признаком не только этой патологии, но и некоторых других генетических заболеваний и мутаций, нарушения синтеза белка. 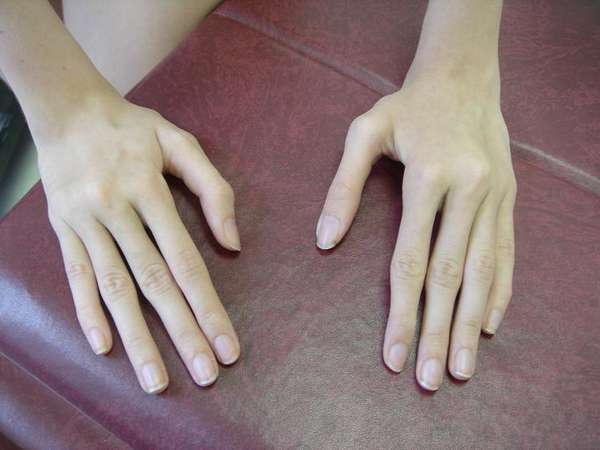
Причины появления
Достоверные и точные причины появления этой патологии у человека до сих пор неизвестны. Большая часть исследователей придерживается той версии, что заболевание связано с нарушением строения коллагеновых волокон и на 75% передается наследственным путем.
Остальные 25% связывают с различными генетическими мутациями, которые могут произойти спонтанно в период зачатия как в яйцеклетке, так и в сперматозоиде.
Формы проявления заболевания, общие симптомы и признаки
Выделяют три основных вида патологических нарушений, для которых характерен симптом «,паучьих пальцев»,:
- Синдром Марфана. Основной вид патологии, передающийся по наследству по аутосомно-доминантному признаку. Сопровождается ярко выраженной деформацией костно-мышечной системы и осложнениями на сердечно-сосудистой системе.
- Гомоцистинурия. Генетическое заболевание, которое передается по аутосомно-рецессивному типу. Редкая патология, встречается примерно у одного человека на 200-300 тысяч.
- Непредвиденные генные мутации, вызывающие патологии различного характера.
Внешние симптомы больного арахнодактилией:
- длинные и тонкие конечности и пальцы,
- выраженный сколиоз, кифосколиоз,
- воронкообразная, суженная, удлиненная грудная клетка,
- талия, расположенная выше нормы,
- аномально высоко сформированная надколенная чашечка,
- деформированные кости таза,
- узкий удлиненный череп,
- неразвитая мышечная система, выраженная худоба,
- широко расставленные глаза,
- высокий свод неба,
- неправильное развитие глаз и другие симптомы.
Основные признаки болезни:
- многочисленные болезни клапанов сердца и аорты,
- гипоплазия легких,
- офтальмологические заболевания (близорукость, косоглазие, катаракта, подвывих хрусталика и др.),
- поражение артерий,
- остеопороз,
- разболтанность суставов,
- припадки,
- могут присутствовать отклонения в психике.
Интересно! Характерный для арахнодактилии выброс адреналина способствует развитию выдающихся умственных способностей и одаренности. Некоторые больные могут отличаться высокой силой духа и сильными волевыми качествами.
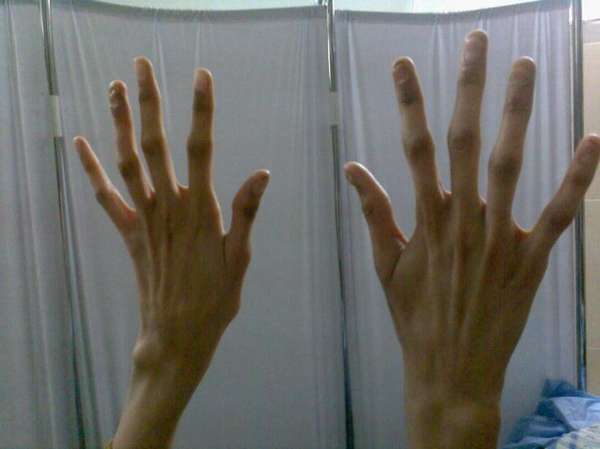
Методы диагностики
Внешние проявления заболевания легко диагностируются при помощи визуального осмотра пациента, замера длины конечностей и гибкости суставов. При обнаружении первых признаков назначается комплексное обследование для выявления сопутствующих заболеваний, патологических изменений, нарушений работы внутренних органов.
Для этого применяют следующие исследования:
- рентген позвоночника, груди и таза,
- МРТ, КТ костей и суставов,
- сцинтиграфия.
Для выявления нарушений работы сердечно-сосудистой системы назначают:
- УЗИ сердца и сосудов,
- томографию артерий и аорты,
- анализы крови.
При подозрении на гомоцистинурию проводят:
- МРТ головного мозга,
- КТ черепа и позвоночника,
- электроэнцефалографию,
- реоэнцефалографию.
Также необходимо обследовать органы зрения у офтальмолога, для чего проводят различные оптические и УЗИ-исследования.
Лечение
Лечение для каждого больного арахнодактилией подбирается строго индивидуально. Это связано с тем, что наличие симптомов или патологий проявляется по-разному. План лечебных и профилактических мероприятий подбирается в зависимости от особенностей протекания болезни. Как правило, основой становятся ортопедические мероприятия, направленные на коррекцию всех имеющихся деформаций.
Полностью устранить болезнь на сегодняшний день нельзя, но есть возможность значительно облегчить состояние больного.
В настоящее время разработан метод коллагенонормализующей терапии, благодаря которому корректируется метаболизм компонентов соединительной ткани, усиливается регенерация хрящей при хирургическом вмешательстве.
Если у человека проявляются заболевания сердечно-сосудистой системы или глаз, то в соответствии с показаниями назначается необходимая терапия по устранению или облегчению симптомов.
При наличии патологий грудной клетки проводится торакопластика – операция по достижению нормального положения грудной клетки и уменьшению ее размеров.
Медикаменты
В зависимости от симптоматики могут быть назначены разнообразные медикаменты, но основными лекарствами являются антиоксиданты и энерготропные препараты: «,Рибоксин»,, «,Коэнзим»,, «,Пирацетам»,, «,Лимонтар»,, витамины группы В и С.
Вне зависимости от причины возникновения заболевания пациенты находятся на постоянном диспансерном наблюдении, посещают ЛФК и массаж.
Важно! ЛФК при арахнодактилии можно применять только при невыраженных нарушениях опорно-двигательного аппарата и исключительно под наблюдением специалиста.
Массаж

Массажные процедуры при арахнодактилии назначаются индивидуально в зависимости от особенностей и патологий пациента, проводятся в комплексе и направлены на лечение и облегчение симптомов патологии.
Массаж выполняется только специально обученными медицинскими работниками, имеющими соответствующую квалификацию.
Заключение
Арахнодактилия — тяжелое и непредсказуемое заболевание, причины появления которого до сих пор не изучены до конца. Чтобы жить с такой патологией, понадобится пройти немало испытаний, проявить силу духа и терпение.
Помимо описанных в статье методов лечения, больному необходимо в качестве профилактики и повышения жизненного тонуса нормализовать сон и обязательно соблюдать все рекомендации врача по питанию и физической активности.
Синдром (болезнь) Марфана
Что это за болезнь?
Синдром Марфана – это наследственное заболевание, передающееся по аутосомно-доминантному типу и характеризуется поражением соединительной ткани и ее компонентов.
Болезнь Марфана вызывается мутированием гена, кодирующего фибриллин -1.
Люди с синдромом Марфана имеют удлиненные конечности, паукообразные пальцы и слабый (недоразвитый) подкожно-жировой слой и сверхгибкие суставы (см. фото ниже).
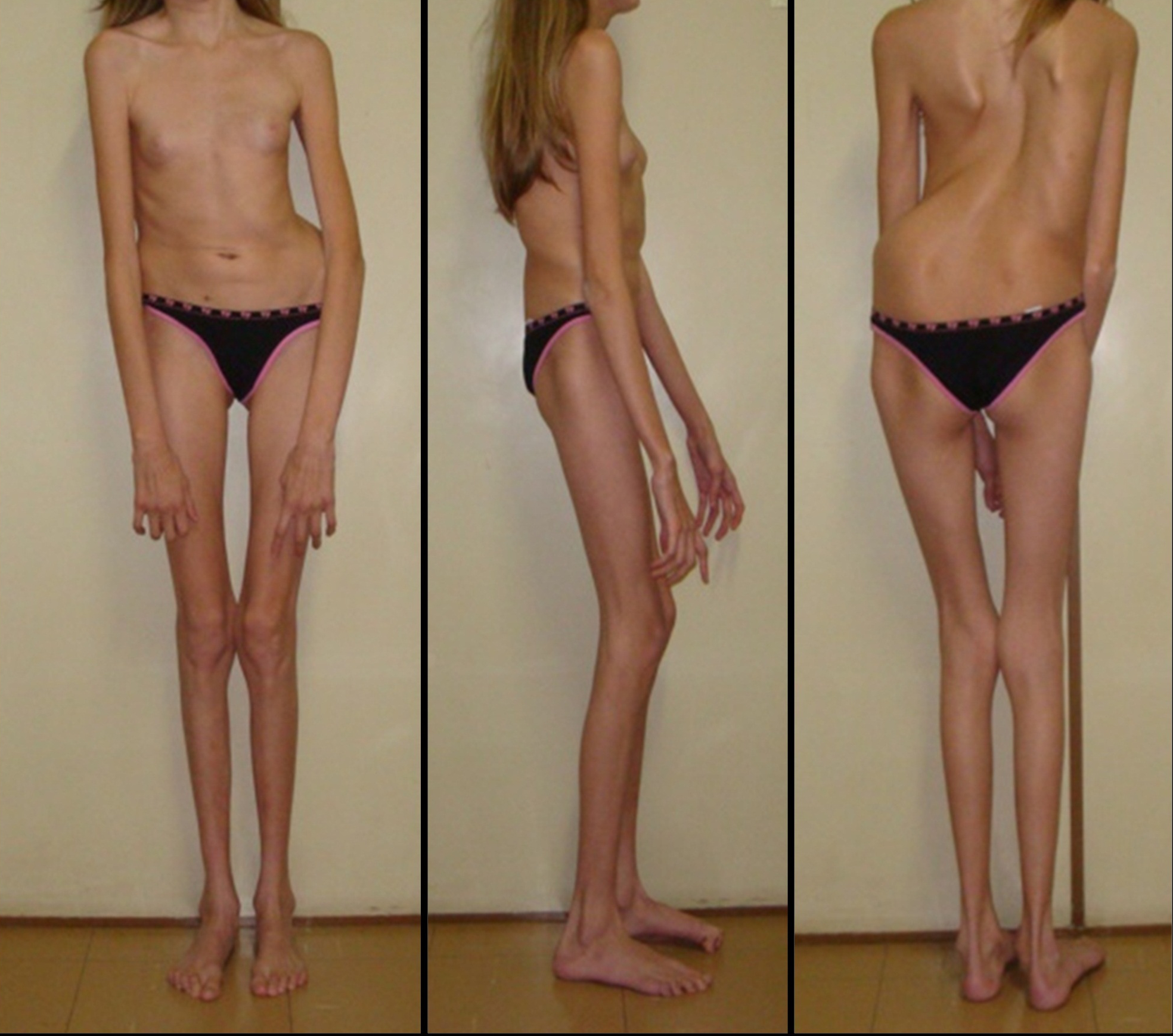
Кроме изменений костно-суставной системы, характерны изменения зрительного анализатора и сердечно-сосудистой системы. Также возможно поражение нервной, дыхательной и других систем.
Впервые описал данную патологию Вильямс, который заметил у своих брата и сестры – выпадение хрусталика, при этом они были очень высокими и имели гипермобильные суставы. Затем заметил Марфан, врач – невролог, у которого в течение 20 лет наблюдалась женщина с подобными симптомами, а затем еще 20 детей.
Причины возникновения

Болезнь Марфана у детей наследуется по аутосомно-доминантному типу (т. е передается от родителя к ребенку).
Также возможны мутации за счет воздействия на организм женщины факторов внешней среды (ионизирующее излучение, лучевая терапия, радиация).
Причины возникновения и механизм развития заболевания недостаточно изучены.
Особая роль отводится нарушению процессов обмена, в результате которых накапливается в коллагеновых и эластических волокнах большое количество мукополисахаридов.
Это ведет к тому, что соединительная ткань перерастягивается, легко подвергается механическому воздействию и приводит к развитию клинической симптоматики.
Классификация
Выделяют следующие формы болезни Марфана:
В зависимости от генной предрасположенности:
- семейная (патология передается от родителя к ребенку);
- спорадическая (патология вызвана внезапным мутированием в геноме).
В зависимости от проявлений клиники:
- стертая, когда признаки заболевания практически не проявляются и могут быть не замечены в течение всей жизни. Патологические изменения выявляются в одной или двух системах.
- выраженная, когда признаки заболевания касаются двух и более органов и систем (сердце, кости и суставы, легкие, кожа, глаза).
Симптомы болезни Марфана
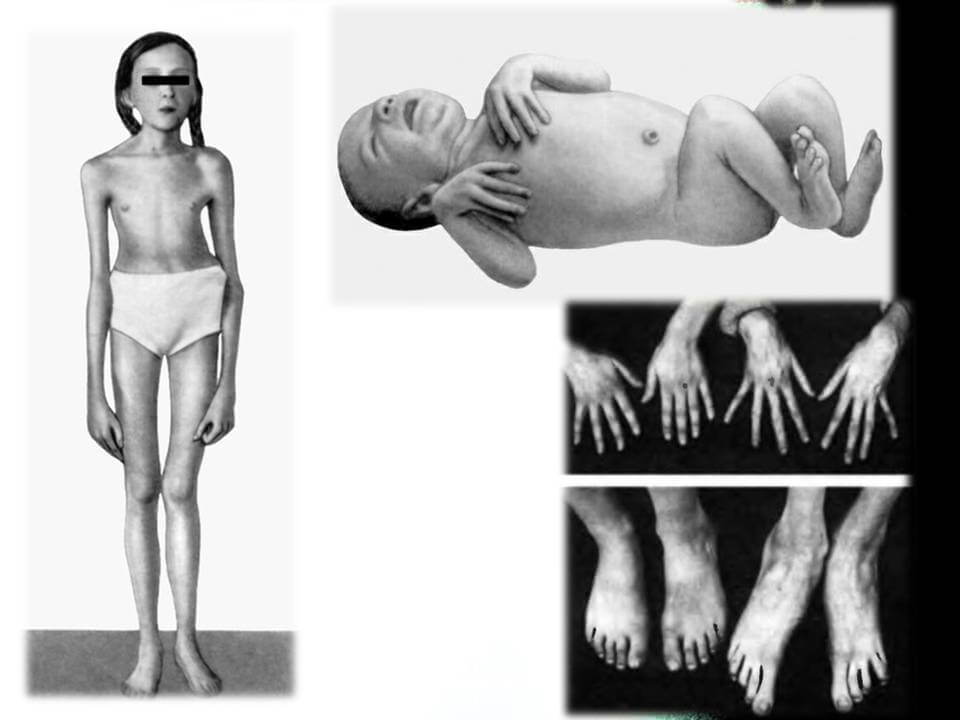
Синдромом Марфана у людей ведет к их выделению в обществе своим непропорциональным строением скелета. Для новорожденных на ранней стадии болезни, характерны длинные пальцы на руках, а к 7-9 годам у детей формируется развернутая клиническая картина.
У взрослых характерна различная симптоматика в зависимости от системы поражения:
- Нервная система: болезненность в поясничной области, головные боли,поражение симпатической и парасимпатической иннервации органов брюшной полости и малого таза (слабость кишечной стенки, недержание мочевого пузыря у ребенка). Также высок риск развития инсульта, субарахноидального кровоизлияния и разрыва аневризм головного мозга.
- Сердечно – сосудистая система: пороки сердца (сужение легочной артерии, пролабирование створок двустворчатого клапана, дилатационная кардиомиопатия, расширение границ сердца (аорты и всех ее отделов), дефекты МЖП и МЖЖ перегородок. У больных может развиваться нарушение ритма и проводимости в виде аритмий.
- Опорно-двигательный аппарат: телосложение астенической формы (дети худые), высокий рост у мужчин 190±10 см, у женщин 179±8 см, слаборазвитый подкожно-жировой слой, длинные пальцы (паукообразные), плоскостопие, череп и лицо вытянутые и узкие, недоразвитость скул, нарушение развития зубов и прикуса, вытянутая нижняя челюсть, готическое верхнее небо, гипермобильность суставов (смотрите на картинки выше). С возрастом ребенка может прогрессировать деформация позвоночного столба, с развитием сколиоза. Также может деформироваться грудная клетка, образуется вдавление – «грудь сапожника». Деформированный тазобедренный сустав нередко приводит к инвалидности при неоказанном своевременном лечении.
- Орган зрения: смещение хрусталика за счет слабого связочного аппарата) на ранней стадии, уплощение роговицы, развитие близорукости или дальнозоркости, спазм аккомодации, отслоение сетчатки.
- Кожа и мягкие ткани: перерастяжение кожи с образованием стрий атрофического характера. Они возникают внезапно, не связаны с колебание веса людей, беременностью и гормональным фоном. Кожа липкая, потная, с мраморным оттенком. Подкожно-жировой слой слабо развит, поэтому у больных наблюдаются грыжевые выпячивания в области передней брюшной стенки.
- Дыхательная система: развитие буллезной эмфиземы легких, проявляющейся кашлем, одышкой, развитием дыхательной недостаточности и спонтанного пневмоторакса.
Другие признаки:
- развитие опущения почек (нефроптоз);
- выпадение органов малого таза (опущение матки у женщин, или ее полное выпадение);
- варикоз вен нижних конечностей;
- запоры.
Диагностика
Диагностика основана на тщательном сборе анамнеза заболевания, выраженности клинической картины, данных осмотра, на результатах лабораторных и инструментальных методов исследований.
Сбор анамнеза включает в себя: наличие в семье данной патологии (родители, братья, сестры) или наличие факторов, провоцирующих мутацию в геноме человека.
К лабораторным методам относят: анализ генотипа ДНК с мутирующим геном, определение гликозаминогликанов в моче.
К инструментальным методам исследования относят:
- ЭКГ, служит для обнаружения патологии сосудов и сердца (ССС). Выявляют характерные нарушения ритма и проводимости в виде мерцательной аритмии, желудочковой экстрасистолии, развитие дилатационной гипертрофии миокарда левого желудочка.
- ЭхоКГ, также служит для обнаружения патологии ССС. Выявляют расширение аорты и ее структур, пролабирование двустворчатого клапана, увеличение размеров левой половины сердца.
- УЗИ сердца проводится для определения осложнений (расслаивающаяся аневризма).
- Рентген органов грудной клетки (изменения скелета, расширение полостей сердца, корней легких и др.)
- Компьютерная томография, магнитно-резонансно-ядерная томография позволяет выявить патологии костно-суставной, нервной системы, нарушение кровообращения в сосудах головного и спинного мозга.
Данные методы исследования служат для обнаружения критериев синдрома Марфана в различных органах и системах. Они играют самую важную роль для постановки и подтверждения диагноза, а в последующем, для определения тактики лечения.
Существуют следующие критерии диагностики синдрома Марфана:
| Система | Большие критерии | Малые критерии |
| Опорно- |
Должны быть: 4 больших критерия, либо 2 больших и 1 малый.
- Грудная клетка неправильной формы: в виде киля / воронки;
- Тесты запястий и большого пальца должны быть положительными;
- Сколиоз;
- Сниженное разгибание локтевых суставов;
- Плоскостопие;
- Выбухание тазобедренного сустава.
- Воронкообразная грудная клетка;
- Переразгибание суставов;
- Готическое нёбо и изменение зубов;
- Изменения лицевого черепа (уплощение).
Для постановки диагноза «Синдром Марфана» учитывается один признак из перечня больших критериев или малый критерий, характерный каждой из пораженной систем, кроме опорно-двигательного аппарата, (необходимо, как минимум, 4 критерия), а также наличие в семейном анамнезе больных с данной патологией.
Лечение синдрома Марфана
От синдрома Марфана полностью избавиться и устранить механизм его развития невозможно. Лечение базируется на улучшении общего состояния больного, устранении клинических проявлений и проведении профилактических мероприятий, препятствующих развитию осложнений.
Больным с данным синдромам рекомендовано ограничить физическую нагрузку до низкого уровня, или минимального. Риск появления патологии сердечно-сосудистой системы возрастает при средних и высоких физических нагрузках.
Следует обходить стороной и повседневные нагрузки, при которых возможно повышение внутригрудного давления, ведущему к развитию пневмоторакса (например, подъем тяжестей, подъем по этажам).
Больные с синдромом Марфана должны консультироваться у разных специалистов, в зависимости от клинически пораженных систем органов. Следует проходить медицинские осмотры каждые полгода в течение всей жизни.
Медикаментозное лечение
Медикаментозная терапия направлена на устранение клинической картины заболевания.
Со стороны сердечно-сосудистой системы рекомендованы β-адреноблокаторы (например: Анаприлин), которые снижают скорость распространения пульсовых волн при стремительно растущем расширении аорты и обратного тока крови на двустворчатом клапане или клапане аорты.
β-адреноблокаторы также оказывают положительный эффект при нарушении ритма и проводимости, в сочетании с сердечными гликозидами.
Но следует помнить о существующих противопоказаниях данных групп препаратов:
- хронический обструктивный бронхит;
- бронхиальная астма;
- снижение частоты сердечных сокращений;
- низкое артериальное давление.
Блокаторы каналов кальция используются при наличии противопоказаний к В-адреноблокаторам.
Хирургия
Хирургическое лечение проводится, если есть осложнения со стороны сердечно-сосудистой системы, с целью коррекции пораженных участков. Его проводят при пролабировании двустворчатого клапана и расслаивании аорты.
При этом осуществляется протезирование двустворчатого клапана.
У беременных с тяжелым течением болезни Марфана, роды разрешаются хирургическим путем.
Профилактика
С профилактической целью, во избежание развития инфекционных осложнений, образования тромбов и тромбоэмболии, назначаются антикоагулянты (гепарин), антибактериальная терапия и витаминотерапия.
- При синдроме Марфана с тяжелым поражением зрительного анализатора проводится хирургическая коррекция зрения, после которой пациенты должны носить очки или контактные линзы.
- Если возникают осложнения, проводят лазерную коррекцию глаукомы, катаракты, удаляют смещаемый хрусталик, заменяя его на искусственный.
- При функциональной дисфункции опорно-двигательного аппарата возникает необходимость стабилизирования позвоночника с помощью металлических пластин.
- При выраженной деформировании грудной клетки проводится торакопластика.
- При протрузии тазобедренных суставов производят внутреннее протезирование суставов.
Прогноз
Длительность жизни в среднем при синдроме Марфана составляет 30-45 лет.
Известно, что это многие знаменитые личности страдали данным синдромом. Это и Ганс Христиан Андерсен – датский писатель, автор знаменитой Русалочки; Авраам Линкольн – 16 президент США, Майкл Феллпс- известный пловец, многократный олимпийский чемпион. А также известные композиторы – Никколо Паганини, Сергей Рахманинов.
Люди с данной патологией должны тщательно следить за своим здоровьем, постоянно наблюдаться и консультироваться со своим лечащим врачом, избегать чрезмерных физических нагрузок.
По мимо медикаментозного лечения, необходимо проведение профилактических мероприятий с целью улучшения общего самочувствия, повышения иммунитета, соответствующий режим труда и отдыха.
Видеозаписи по теме
Как лечить паучьи пальцы
В числе серьезных генетических заболеваний находится синдром Марфана, которым страдает один человек из 10-20 тысяч. Им болели такие всемирно известные личности, как Авраам Линкольн, Никколо Паганини, Корней Чуковский и Ганс Христиан Андерсен.
У больного этим синдромом присутствуют различные симптомы: арахнодактилия (удлиненные кисти, паукообразные пальцы рук и ног), множественные костно-суставные аномалии, деформация грудной клетки, нарушения сердечно-сосудистой системы и т.д. В этой статье речь пойдет об одном из них — арахнодактилии, ее проявлениях, методах диагностики и способах лечения.
Что такое арахнодактилия
Это вид наследственной патологии человека, который проявляется аномально длинными и тонкими пальцами рук, их чрезмерной подвижностью, вплоть до сгибания на 180 градусов к тыльной стороне ладони. В народе это заболевание приобрело название болезни «,паучьи пальцы»,.
После описанного впервые синдрома Марфана в 1876 году было выявлено, что «,паучьи пальцы», могут быть признаком не только этой патологии, но и некоторых других генетических заболеваний и мутаций, нарушения синтеза белка.
Причины появления
Достоверные и точные причины появления этой патологии у человека до сих пор неизвестны. Большая часть исследователей придерживается той версии, что заболевание связано с нарушением строения коллагеновых волокон и на 75% передается наследственным путем.
Остальные 25% связывают с различными генетическими мутациями, которые могут произойти спонтанно в период зачатия как в яйцеклетке, так и в сперматозоиде.
Формы проявления заболевания, общие симптомы и признаки
Выделяют три основных вида патологических нарушений, для которых характерен симптом «,паучьих пальцев»,:
- Синдром Марфана. Основной вид патологии, передающийся по наследству по аутосомно-доминантному признаку. Сопровождается ярко выраженной деформацией костно-мышечной системы и осложнениями на сердечно-сосудистой системе.
- Гомоцистинурия. Генетическое заболевание, которое передается по аутосомно-рецессивному типу. Редкая патология, встречается примерно у одного человека на 200-300 тысяч.
- Непредвиденные генные мутации, вызывающие патологии различного характера.
Внешние симптомы больного арахнодактилией:
- длинные и тонкие конечности и пальцы,
- выраженный сколиоз, кифосколиоз,
- воронкообразная, суженная, удлиненная грудная клетка,
- талия, расположенная выше нормы,
- аномально высоко сформированная надколенная чашечка,
- деформированные кости таза,
- узкий удлиненный череп,
- неразвитая мышечная система, выраженная худоба,
- широко расставленные глаза,
- высокий свод неба,
- неправильное развитие глаз и другие симптомы.
Основные признаки болезни:
- многочисленные болезни клапанов сердца и аорты,
- гипоплазия легких,
- офтальмологические заболевания (близорукость, косоглазие, катаракта, подвывих хрусталика и др.),
- поражение артерий,
- остеопороз,
- разболтанность суставов,
- припадки,
- могут присутствовать отклонения в психике.
Интересно! Характерный для арахнодактилии выброс адреналина способствует развитию выдающихся умственных способностей и одаренности. Некоторые больные могут отличаться высокой силой духа и сильными волевыми качествами.
Методы диагностики
Внешние проявления заболевания легко диагностируются при помощи визуального осмотра пациента, замера длины конечностей и гибкости суставов. При обнаружении первых признаков назначается комплексное обследование для выявления сопутствующих заболеваний, патологических изменений, нарушений работы внутренних органов.
Для этого применяют следующие исследования:
- рентген позвоночника, груди и таза,
- МРТ, КТ костей и суставов,
- сцинтиграфия.
Для выявления нарушений работы сердечно-сосудистой системы назначают:
- УЗИ сердца и сосудов,
- томографию артерий и аорты,
- анализы крови.
При подозрении на гомоцистинурию проводят:
- МРТ головного мозга,
- КТ черепа и позвоночника,
- электроэнцефалографию,
- реоэнцефалографию.
Также необходимо обследовать органы зрения у офтальмолога, для чего проводят различные оптические и УЗИ-исследования.
Лечение
Лечение для каждого больного арахнодактилией подбирается строго индивидуально. Это связано с тем, что наличие симптомов или патологий проявляется по-разному. План лечебных и профилактических мероприятий подбирается в зависимости от особенностей протекания болезни. Как правило, основой становятся ортопедические мероприятия, направленные на коррекцию всех имеющихся деформаций.
Полностью устранить болезнь на сегодняшний день нельзя, но есть возможность значительно облегчить состояние больного.
В настоящее время разработан метод коллагенонормализующей терапии, благодаря которому корректируется метаболизм компонентов соединительной ткани, усиливается регенерация хрящей при хирургическом вмешательстве.
Если у человека проявляются заболевания сердечно-сосудистой системы или глаз, то в соответствии с показаниями назначается необходимая терапия по устранению или облегчению симптомов.
При наличии патологий грудной клетки проводится торакопластика – операция по достижению нормального положения грудной клетки и уменьшению ее размеров.
Медикаменты
В зависимости от симптоматики могут быть назначены разнообразные медикаменты, но основными лекарствами являются антиоксиданты и энерготропные препараты: «,Рибоксин»,, «,Коэнзим»,, «,Пирацетам»,, «,Лимонтар»,, витамины группы В и С.
Вне зависимости от причины возникновения заболевания пациенты находятся на постоянном диспансерном наблюдении, посещают ЛФК и массаж.
Важно! ЛФК при арахнодактилии можно применять только при невыраженных нарушениях опорно-двигательного аппарата и исключительно под наблюдением специалиста.
Массаж
Массажные процедуры при арахнодактилии назначаются индивидуально в зависимости от особенностей и патологий пациента, проводятся в комплексе и направлены на лечение и облегчение симптомов патологии.
Массаж выполняется только специально обученными медицинскими работниками, имеющими соответствующую квалификацию.
Заключение
Арахнодактилия — тяжелое и непредсказуемое заболевание, причины появления которого до сих пор не изучены до конца. Чтобы жить с такой патологией, понадобится пройти немало испытаний, проявить силу духа и терпение.
Помимо описанных в статье методов лечения, больному необходимо в качестве профилактики и повышения жизненного тонуса нормализовать сон и обязательно соблюдать все рекомендации врача по питанию и физической активности.